Vous aimerez peut-être aussi
- Combined Lab ReportDocument9 pagesCombined Lab Reportapi-27051220550% (2)
- Size Exclusive ChromatographyDocument7 pagesSize Exclusive Chromatographygrant1115Pas encore d'évaluation
- Sutherland 1991Document7 pagesSutherland 1991Isal AbdussalamPas encore d'évaluation
- Lab Report Sds-Page WB - PT 1 (1-5)Document5 pagesLab Report Sds-Page WB - PT 1 (1-5)Ezad juferiPas encore d'évaluation
- DNA Quality-Spectrophotometry and ElectrophoresisDocument5 pagesDNA Quality-Spectrophotometry and Electrophoresislovina candra kirana100% (1)
- Introduction MTT AssayDocument10 pagesIntroduction MTT Assay16_dev5038Pas encore d'évaluation
- Restriction Enzyme DigestionDocument5 pagesRestriction Enzyme DigestionAqsa ImtiazPas encore d'évaluation
- Restriction Enzyme Digestion AnalysisDocument6 pagesRestriction Enzyme Digestion AnalysisLloaana 12Pas encore d'évaluation
- Protein TechniquesDocument13 pagesProtein TechniquesRendel GuevarraPas encore d'évaluation
- Protein Extraction From AlgaeDocument8 pagesProtein Extraction From AlgaecookooponyPas encore d'évaluation
- Gel FiltrationDocument123 pagesGel FiltrationZulfikri Asmardi RaufPas encore d'évaluation
- Most Effective Lipid Extraction Method from MicroalgaeDocument3 pagesMost Effective Lipid Extraction Method from MicroalgaeRahul GogiaPas encore d'évaluation
- Gel FiltrationDocument5 pagesGel FiltrationRüveyda AkçinPas encore d'évaluation
- Quantitative Determination of Proteins Using Bradford MethodDocument2 pagesQuantitative Determination of Proteins Using Bradford Methodann_michelle7Pas encore d'évaluation
- EXP5CHEM26Document12 pagesEXP5CHEM26Albert Romano ObisPas encore d'évaluation
- Dna StructureDocument23 pagesDna StructureAnonymous mHS76aPas encore d'évaluation
- DNA ExtractionDocument18 pagesDNA ExtractionUmmi MahmudahPas encore d'évaluation
- A Potassium Manganate Vii Ammonium Iron II Sulfate TitrationDocument5 pagesA Potassium Manganate Vii Ammonium Iron II Sulfate TitrationChong Fei0% (1)
- Chem 160.1 Post LabDocument7 pagesChem 160.1 Post LabMarlyn AmantePas encore d'évaluation
- Gram Stain Prac ReportDocument4 pagesGram Stain Prac ReportToga BrandonPas encore d'évaluation
- Energy Metabolism in The Liver: Liangyou RuiDocument21 pagesEnergy Metabolism in The Liver: Liangyou Ruidwi yuniariPas encore d'évaluation
- Bacterial Transformation Lab (6a)Document7 pagesBacterial Transformation Lab (6a)Chris PricePas encore d'évaluation
- Production, Purification and Assay of Pectinase Enzyme From Aspergillus NigerDocument6 pagesProduction, Purification and Assay of Pectinase Enzyme From Aspergillus NigerEE KMPas encore d'évaluation
- UV Absorbance: Click To Edit Master Subtitle StyleDocument24 pagesUV Absorbance: Click To Edit Master Subtitle StylePaula Denice Carlos BagunuPas encore d'évaluation
- Lab Report Exp.6Document8 pagesLab Report Exp.6Qj B PdkhPas encore d'évaluation
- Experiment 1 (Introduction)Document16 pagesExperiment 1 (Introduction)Msfaeza HanafiPas encore d'évaluation
- The Bradford Method For Protein QuantitationDocument7 pagesThe Bradford Method For Protein QuantitationChemiboyPas encore d'évaluation
- Calculations TutorialDocument9 pagesCalculations Tutorialricara alexia moodley0% (1)
- Purification and Citrate Inhibition of Fumarase From YeastDocument1 pagePurification and Citrate Inhibition of Fumarase From YeastzarobannPas encore d'évaluation
- 2-14 Epoxidation of AlkenesDocument2 pages2-14 Epoxidation of AlkenesMohd Zulhelmi AzmiPas encore d'évaluation
- DNA Extraction Reagents - FunctionsDocument1 pageDNA Extraction Reagents - Functionsharpreet157100% (5)
- Amino Acid Titration Experiment Determines Acid-Base BehaviorDocument5 pagesAmino Acid Titration Experiment Determines Acid-Base BehaviorNatalie Cu100% (1)
- S.Y.Bsc Semester Iii Botany Paper IiDocument53 pagesS.Y.Bsc Semester Iii Botany Paper IiĐỗ Quang BìnhPas encore d'évaluation
- BIOL 3140 Lab Report 3Document21 pagesBIOL 3140 Lab Report 3Tabashir AhmedPas encore d'évaluation
- SDS PageDocument5 pagesSDS Pagestevensb055Pas encore d'évaluation
- DNA Isolation, Restriction, Visualitation, and QuantificationDocument20 pagesDNA Isolation, Restriction, Visualitation, and QuantificationSonianto kuddi100% (5)
- Efficient Reduction of Imines to Secondary AminesDocument4 pagesEfficient Reduction of Imines to Secondary AminesRatna Siti KhodijahPas encore d'évaluation
- Triple Sugar Iron AgarDocument3 pagesTriple Sugar Iron AgarmaniPas encore d'évaluation
- Plasmid IsolationDocument4 pagesPlasmid IsolationSindhu LakshmiPas encore d'évaluation
- Bradford Formal ReportDocument4 pagesBradford Formal ReportAlyana100% (1)
- BCH 314 Tutorial 1 SolutionsDocument9 pagesBCH 314 Tutorial 1 SolutionsvictorPas encore d'évaluation
- 2016 BCH223 PracticalsDocument7 pages2016 BCH223 PracticalsvictorPas encore d'évaluation
- BI309 Practical 6Document8 pagesBI309 Practical 6SanahKumar100% (1)
- Protein Extraction Buffer ComponentsDocument1 pageProtein Extraction Buffer ComponentsRabia AnjumPas encore d'évaluation
- Estimation of DNADocument1 pageEstimation of DNATjcbt BiosciencesPas encore d'évaluation
- Tyra Nelson FINAL Total Glycated HaemoglobinDocument7 pagesTyra Nelson FINAL Total Glycated HaemoglobinK WPas encore d'évaluation
- SDS-PAGE Guide: Separate Proteins by Size & ChargeDocument39 pagesSDS-PAGE Guide: Separate Proteins by Size & ChargeSnehalphirke100% (1)
- H&E Staining Overview: A Guide To Best Practices:, JD CT (Ascp) HTL, Bappsc, FaimsDocument29 pagesH&E Staining Overview: A Guide To Best Practices:, JD CT (Ascp) HTL, Bappsc, FaimsAnonymous iqoU1mtPas encore d'évaluation
- Purification of DNADocument14 pagesPurification of DNAalivetutorsPas encore d'évaluation
- QUESTION 1 (52 Marks) : Biochemistry 3 BCH 314Document4 pagesQUESTION 1 (52 Marks) : Biochemistry 3 BCH 314victorPas encore d'évaluation
- Size Exclusion Column ChromatographyDocument8 pagesSize Exclusion Column ChromatographySmeetha Kaur100% (1)
- Biochem 313 Prac 5Document8 pagesBiochem 313 Prac 5Anonymous G8WVOfRqV100% (2)
- Cell ViabilityTesting With Trypan Blue Exclusion MethodDocument2 pagesCell ViabilityTesting With Trypan Blue Exclusion MethodRajeev PotadarPas encore d'évaluation
- BiochemistryDocument5 pagesBiochemistryAngeline LimpiadaPas encore d'évaluation
- Types of PCRDocument27 pagesTypes of PCRnandhiniPas encore d'évaluation
- Isolation and Enumeration of Bacteria in Water and FoodDocument30 pagesIsolation and Enumeration of Bacteria in Water and FoodOld Lake100% (1)
- Staining TechniquesDocument19 pagesStaining TechniquesSwayamprakash PatelPas encore d'évaluation
- The Pyridine Nucleotide CoenzymesD'EverandThe Pyridine Nucleotide CoenzymesJohannes EversePas encore d'évaluation
- Proposal PresentationDocument11 pagesProposal Presentationapi-283089766Pas encore d'évaluation
- Proposal Presentation EssayDocument7 pagesProposal Presentation Essayapi-283089766Pas encore d'évaluation
- Phys2125 Lab ReportDocument6 pagesPhys2125 Lab Reportapi-283089766Pas encore d'évaluation
- Resume FinalDocument1 pageResume Finalapi-283089766Pas encore d'évaluation
- PHA6112 - Lab - ProteinsDocument16 pagesPHA6112 - Lab - ProteinsPompeyo BarrogaPas encore d'évaluation
- Protein Isolation TechniquesDocument77 pagesProtein Isolation TechniquesEricka PutriPas encore d'évaluation
- International Biodeterioration & BiodegradationDocument8 pagesInternational Biodeterioration & Biodegradationchristopher cordova gutierrezPas encore d'évaluation
- The Bradford Method For Protein Quantitation. Nicholas Kruger PDFDocument7 pagesThe Bradford Method For Protein Quantitation. Nicholas Kruger PDFGise Clara HernandezPas encore d'évaluation
- Biochemistry Lab ManualDocument40 pagesBiochemistry Lab Manualharpreet0% (1)
- Proteomic ReportDocument37 pagesProteomic ReportNaina Karamina SakinaPas encore d'évaluation
- Experiment 4: Protein Assay SpectrophotometryDocument9 pagesExperiment 4: Protein Assay SpectrophotometryShadia HeyariPas encore d'évaluation
- Protein Concentration Estimation Using Bradford AssayDocument2 pagesProtein Concentration Estimation Using Bradford AssayMarc DacumosPas encore d'évaluation
- Bradford Protein Cuantification AssayDocument2 pagesBradford Protein Cuantification AssayFabio Palacios100% (1)
- 200-Protein Quantification BCA™, Modified Lowry and Bradford AssaysDocument5 pages200-Protein Quantification BCA™, Modified Lowry and Bradford AssaysMusa LooPas encore d'évaluation
- Fermentation LabDocument12 pagesFermentation LabChiPas encore d'évaluation
- Analytical Methods: PaperDocument8 pagesAnalytical Methods: PaperkaltoumPas encore d'évaluation
- The Bradford Method For Protein QuantitationDocument7 pagesThe Bradford Method For Protein QuantitationChemiboyPas encore d'évaluation
- Lowry's Method of Protein EstimationDocument21 pagesLowry's Method of Protein EstimationSwetha Sundar100% (1)
- Lonza ManualsProductInstructions Determination of Protein Concentration 31460Document2 pagesLonza ManualsProductInstructions Determination of Protein Concentration 31460sydneypadillioPas encore d'évaluation
- Summary of Bradford Assay: Fig: Chemical Structures of Coomassie Brilliant Blue G-250Document2 pagesSummary of Bradford Assay: Fig: Chemical Structures of Coomassie Brilliant Blue G-250Pradeep Subedi100% (1)
- Assays For Determination of Protein Concentration PDFDocument32 pagesAssays For Determination of Protein Concentration PDFImanol Cuevas Medina100% (1)
- Protein quantification using Bradford methodDocument2 pagesProtein quantification using Bradford methodAly Arcega100% (4)
- Bradford Formal ReportDocument4 pagesBradford Formal ReportAlyana100% (1)
- Spectrophotometric Determination of Protein ConcentrationDocument16 pagesSpectrophotometric Determination of Protein ConcentrationTouhid IslamPas encore d'évaluation
- Ion-Exchange and Gel Filtration Chromatography for Protein PurificationDocument5 pagesIon-Exchange and Gel Filtration Chromatography for Protein PurificationJoPas encore d'évaluation
- Dilution and Molarity ExperimentsDocument5 pagesDilution and Molarity Experimentsfuyuki miharuPas encore d'évaluation
- BIOCHEMDocument2 pagesBIOCHEMharita_ludba89100% (6)
- 158an Bradford Protein ConcentrationDocument2 pages158an Bradford Protein ConcentrationBIO45Pas encore d'évaluation
- Quantitation of Protein: James E. Noble and Marc J. A. BaileyDocument23 pagesQuantitation of Protein: James E. Noble and Marc J. A. BaileyChasconaPas encore d'évaluation
- Separation and Identification of Myoglobin by Paper Chromatography and Protein Assay by Bradford MethodDocument6 pagesSeparation and Identification of Myoglobin by Paper Chromatography and Protein Assay by Bradford MethodJason Montesa100% (1)
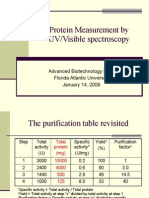
- Protein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Document10 pagesProtein Measurement by UV/Visible Spectroscopy: Advanced Biotechnology Lab I Florida Atlantic University January 14, 2008Gina ZhouPas encore d'évaluation
- Food Analysis Chapter 18Document8 pagesFood Analysis Chapter 18ngoc.nguyenlamPas encore d'évaluation
- Bradford Method Protein Assay GuideDocument3 pagesBradford Method Protein Assay GuideDoreliaPas encore d'évaluation
- Quantitative protein analysis: Determining protein concentration using Bradford assayDocument3 pagesQuantitative protein analysis: Determining protein concentration using Bradford assaySean Herman100% (1)