Vous aimerez peut-être aussi
- Beyond Compliance Design of a Quality System: Tools and Templates for Integrating Auditing PerspectivesD'EverandBeyond Compliance Design of a Quality System: Tools and Templates for Integrating Auditing PerspectivesPas encore d'évaluation
- Data Integrity and Compliance: A Primer for Medical Product ManufacturersD'EverandData Integrity and Compliance: A Primer for Medical Product ManufacturersPas encore d'évaluation
- Eu MDRDocument34 pagesEu MDRgobu269104100% (1)
- ISO 14971 A Complete Guide - 2021 EditionD'EverandISO 14971 A Complete Guide - 2021 EditionÉvaluation : 1 sur 5 étoiles1/5 (1)
- Eu GMP Guide PDFDocument2 pagesEu GMP Guide PDFSteve0% (1)
- Risk Management for Medical Device Manufacturers: [MD and IVD]D'EverandRisk Management for Medical Device Manufacturers: [MD and IVD]Pas encore d'évaluation
- GHTF Supplier Controlsg3final N17Document21 pagesGHTF Supplier Controlsg3final N17freelovefestPas encore d'évaluation
- ISO 13485 Quality Management System A Complete Guide - 2020 EditionD'EverandISO 13485 Quality Management System A Complete Guide - 2020 EditionPas encore d'évaluation
- Achieving 6 Sigma Quality in Medical Device Manufacturing by Use of DoE and SPCDocument9 pagesAchieving 6 Sigma Quality in Medical Device Manufacturing by Use of DoE and SPCispam28Pas encore d'évaluation
- GHTF Sg3 n99 8 Guidance On Quality 990629Document48 pagesGHTF Sg3 n99 8 Guidance On Quality 990629BALAJIPas encore d'évaluation
- SS ISO 10993-1-2018 - PreviewDocument14 pagesSS ISO 10993-1-2018 - PreviewmarkPas encore d'évaluation
- Good Distribution Practices A Complete Guide - 2021 EditionD'EverandGood Distribution Practices A Complete Guide - 2021 EditionPas encore d'évaluation
- Design Documentation L4Document27 pagesDesign Documentation L4KOFI BROWNPas encore d'évaluation
- Capa-UpDocument22 pagesCapa-UpChristinaPas encore d'évaluation
- Overview PF CPGPDocument26 pagesOverview PF CPGPmmmmm0% (1)
- How Medical Devices Regulated in EuropeDocument7 pagesHow Medical Devices Regulated in Europebasakerpolat100% (1)
- Ord 384-2020 - ENGLISCHDocument54 pagesOrd 384-2020 - ENGLISCHScribdTranslationsPas encore d'évaluation
- Quality Management System Software A Complete Guide - 2019 EditionD'EverandQuality Management System Software A Complete Guide - 2019 EditionPas encore d'évaluation
- HPS Endoscope Decontamination 2004 PDFDocument190 pagesHPS Endoscope Decontamination 2004 PDFHong-Nam Kim100% (1)
- Thermal Validation Information Manual & GuideDocument10 pagesThermal Validation Information Manual & Guideviethuong96Pas encore d'évaluation
- RDC 16 2013 GMP Requirements For MD and IvdDocument12 pagesRDC 16 2013 GMP Requirements For MD and Ivdnsk79inPas encore d'évaluation
- ISO 14971 A Complete Guide - 2020 EditionD'EverandISO 14971 A Complete Guide - 2020 EditionÉvaluation : 2 sur 5 étoiles2/5 (1)
- Australian Regulatory Guidelines For Medical DevicesDocument337 pagesAustralian Regulatory Guidelines For Medical DevicesLina GamarnikPas encore d'évaluation
- FDA Inspections PDFDocument52 pagesFDA Inspections PDFMajdi Hasan Ayoub100% (1)
- Analyzing The Changes To Risk Management Standard ISO 149712019 PDFDocument5 pagesAnalyzing The Changes To Risk Management Standard ISO 149712019 PDFrakesh marwahPas encore d'évaluation
- Sterilization - Validation, Qualification RequirementsDocument2 pagesSterilization - Validation, Qualification Requirementsgloryclaudia_100% (1)
- Validation Vs QualificationDocument1 pageValidation Vs QualificationKiran ChokshiPas encore d'évaluation
- Validation Change Control Maintaining The Validate State-09-2015Document56 pagesValidation Change Control Maintaining The Validate State-09-2015VaLeritha R. Santa MPas encore d'évaluation
- Unit IV - Complaints and DocumentsDocument103 pagesUnit IV - Complaints and DocumentsIndu VattiPas encore d'évaluation
- Quality by Design Approaches To Analytical Methods - : FDA PerspectiveDocument21 pagesQuality by Design Approaches To Analytical Methods - : FDA PerspectiveJr Zdenko VukojaPas encore d'évaluation
- Validation of Microbiology PDFDocument6 pagesValidation of Microbiology PDFMayur Jadhav0% (1)
- As Far As Possible - en ISO 14971Document19 pagesAs Far As Possible - en ISO 14971Kanwal Jit Singh100% (1)
- Basic Principles of GMP Basic Principles of GMPDocument33 pagesBasic Principles of GMP Basic Principles of GMPTahir KhanPas encore d'évaluation
- Basic Principles of GMP: Qualification and ValidationDocument28 pagesBasic Principles of GMP: Qualification and Validationhyde2520015754100% (1)
- Quality Systems For Pharmacovigilance SME Workshop "Focus On Pharmacovigilance" 19 April 2012, EMA LondonDocument20 pagesQuality Systems For Pharmacovigilance SME Workshop "Focus On Pharmacovigilance" 19 April 2012, EMA LondonHala MohamedPas encore d'évaluation
- Manufacturing Facilities A Complete Guide - 2019 EditionD'EverandManufacturing Facilities A Complete Guide - 2019 EditionPas encore d'évaluation
- Study - ISO 13485 PDFDocument15 pagesStudy - ISO 13485 PDFAnonymous 78Ezy46qvPas encore d'évaluation
- PIT Virtual IAI 2020 Presentation - Data Integrity - Silvia WidyanyDocument28 pagesPIT Virtual IAI 2020 Presentation - Data Integrity - Silvia WidyanyAchmad Fadhil AlmasyhurPas encore d'évaluation
- SS IEC 62366-1-2018 PreviewDocument12 pagesSS IEC 62366-1-2018 PreviewMADDINENI AVANEESHWARPas encore d'évaluation
- Standards Version 1.0Document28 pagesStandards Version 1.0maxPas encore d'évaluation
- Biocompatability, FDA, IsO 10993, IVDDocument31 pagesBiocompatability, FDA, IsO 10993, IVDsam3557Pas encore d'évaluation
- FDA Medical Device Quality Systems Manual - Quality Systems AuditsDocument5 pagesFDA Medical Device Quality Systems Manual - Quality Systems AuditsucbdmoPas encore d'évaluation
- Application Note: Successful Wetting For Filter Integrity Testing in Volume-Restricted SystemsDocument13 pagesApplication Note: Successful Wetting For Filter Integrity Testing in Volume-Restricted SystemsSlavaPas encore d'évaluation
- CPGP CourseDocument7 pagesCPGP CourseantonygamalpharmaPas encore d'évaluation
- Quality Data Metrics A Complete Guide - 2020 EditionD'EverandQuality Data Metrics A Complete Guide - 2020 EditionPas encore d'évaluation
- 413-Other Tools-Control PlanDocument8 pages413-Other Tools-Control PlanAnkur DhirPas encore d'évaluation
- Lesson 4: Other Quality Tools Tool #8: BrainstormingDocument4 pagesLesson 4: Other Quality Tools Tool #8: BrainstormingAnkur DhirPas encore d'évaluation
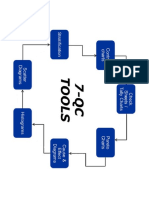
- 7 QC ToolDocument1 page7 QC ToolAnkur DhirPas encore d'évaluation
- 5 S of Lean PosterDocument1 page5 S of Lean PosterAnkur DhirPas encore d'évaluation
- 412-Other Tools-Criteria RatingDocument6 pages412-Other Tools-Criteria RatingAnkur DhirPas encore d'évaluation
- Performance Measurement: Lesson StructureDocument9 pagesPerformance Measurement: Lesson StructureAnkur DhirPas encore d'évaluation
- 410-Other Quality tools-ProcessFlowDocument6 pages410-Other Quality tools-ProcessFlowAnkur DhirPas encore d'évaluation
- Seven QC Tools Tool #7: Stratification: Lesson StructureDocument2 pagesSeven QC Tools Tool #7: Stratification: Lesson StructureAnkur DhirPas encore d'évaluation
- 49-Other Quality Tools-5 WhysDocument4 pages49-Other Quality Tools-5 WhysAnkur DhirPas encore d'évaluation
- Seven QC Tools Tool #5: Part 1-Run ChartDocument6 pagesSeven QC Tools Tool #5: Part 1-Run ChartAnkur DhirPas encore d'évaluation
- Lesson 3: Seven QC Tools Tool #3: Cause & Effect DiagramDocument6 pagesLesson 3: Seven QC Tools Tool #3: Cause & Effect DiagramAnkur DhirPas encore d'évaluation
- Lean Coffee Table Book 8 - 18-5-2k18Document48 pagesLean Coffee Table Book 8 - 18-5-2k18Ankur DhirPas encore d'évaluation
- Seven QC Tools Tool #4: Pareto ChartDocument6 pagesSeven QC Tools Tool #4: Pareto ChartAnkur DhirPas encore d'évaluation
- 1-Problem SolvingDocument8 pages1-Problem SolvingAnkur DhirPas encore d'évaluation
- IRIS Beyond ISO 9001 - HandoutDocument83 pagesIRIS Beyond ISO 9001 - HandoutAnkur DhirPas encore d'évaluation
- 36-7QC Tools-Scatter DiagramDocument6 pages36-7QC Tools-Scatter DiagramAnkur DhirPas encore d'évaluation
- 2-Overview of Quality ToolsDocument3 pages2-Overview of Quality ToolsAnkur DhirPas encore d'évaluation
- Social Responsibility and Performance ExcellenceDocument4 pagesSocial Responsibility and Performance ExcellenceAnkur DhirPas encore d'évaluation
- Readiness Review: ISO RevisionsDocument6 pagesReadiness Review: ISO RevisionsAnkur DhirPas encore d'évaluation
- Session 3 Project Management 2Document15 pagesSession 3 Project Management 2Ankur DhirPas encore d'évaluation
- Best Practices Organizational AgilityDocument23 pagesBest Practices Organizational AgilityAnkur Dhir100% (1)
- Reinventing ExcellenceDocument7 pagesReinventing ExcellenceAnkur DhirPas encore d'évaluation
- Session 1 Office ManagementDocument20 pagesSession 1 Office ManagementAnkur DhirPas encore d'évaluation
- Session 2 Session 2: Introduction To Project Management Introduction To Project ManagementDocument24 pagesSession 2 Session 2: Introduction To Project Management Introduction To Project ManagementAnkur DhirPas encore d'évaluation
- Effective Team SkillsDocument12 pagesEffective Team SkillsAnkur Dhir100% (1)
- Risk Management QMS MethodolgyDocument35 pagesRisk Management QMS MethodolgyAnkur DhirPas encore d'évaluation
- Raci Matrix For Itsms 20000Document3 pagesRaci Matrix For Itsms 20000Ankur DhirPas encore d'évaluation
- Ocp For Fire Fighting Equipment and First Aid BoxesDocument2 pagesOcp For Fire Fighting Equipment and First Aid BoxesAnkur DhirPas encore d'évaluation
- Product Packing Plan: SL NO Item Under Packing Packing Size Weight (In KGS) of Packing Type of PackingDocument1 pageProduct Packing Plan: SL NO Item Under Packing Packing Size Weight (In KGS) of Packing Type of PackingAnkur DhirPas encore d'évaluation
- Synthesis and Characterization of Alumina Zirconia Composite Material Doped With SilicaDocument7 pagesSynthesis and Characterization of Alumina Zirconia Composite Material Doped With SilicaAdvanced Research PublicationsPas encore d'évaluation
- Dr. Ambedkar Institute of Technology: Mandatory Non-Credit NSS Course (22NSN310) Bachelor of Engineering inDocument4 pagesDr. Ambedkar Institute of Technology: Mandatory Non-Credit NSS Course (22NSN310) Bachelor of Engineering inshamalac2004Pas encore d'évaluation
- 4455770110e1 Parts ManualDocument31 pages4455770110e1 Parts ManualSimon AlbertPas encore d'évaluation
- PESTLE Analysis Patanjali Ayurved LTDDocument7 pagesPESTLE Analysis Patanjali Ayurved LTDvaidehi50% (2)
- Modelling and Simulation of Absorption Solar Air Conditioning SystemDocument20 pagesModelling and Simulation of Absorption Solar Air Conditioning SystemShadi KarkabaPas encore d'évaluation
- Building 101 - 25 Tips For A Tropical HomeDocument11 pagesBuilding 101 - 25 Tips For A Tropical HomeMelanie CabforoPas encore d'évaluation
- Whose Online Video Lectures Are Best For The IIT JEE?Document6 pagesWhose Online Video Lectures Are Best For The IIT JEE?Buzz PriyanshuPas encore d'évaluation
- S9300&S9300E V200R001C00 Hardware Description 05 PDFDocument282 pagesS9300&S9300E V200R001C00 Hardware Description 05 PDFmike_mnleePas encore d'évaluation
- 7sd610 CatalogueDocument35 pages7sd610 CatalogueTntngn Petualang100% (1)
- Average - Aptitude MCQ Questions and Solutions Wit - 1597107113795Document6 pagesAverage - Aptitude MCQ Questions and Solutions Wit - 1597107113795Manish ChavannavarPas encore d'évaluation
- Bloomberg Certification FAQ BMC (Bloomberg Market Concepts) : Goizueta Business LibraryDocument3 pagesBloomberg Certification FAQ BMC (Bloomberg Market Concepts) : Goizueta Business LibrarySarah Raquel Bozo Herrera100% (1)
- Trial On CompresorDocument3 pagesTrial On CompresorA JPas encore d'évaluation
- Quick Start Guide: Digital Camera D7000Document2 pagesQuick Start Guide: Digital Camera D7000foosome12Pas encore d'évaluation
- 9500MPR - MEF8 Circuit Emulation ServicesDocument5 pages9500MPR - MEF8 Circuit Emulation ServicesedderjpPas encore d'évaluation
- Poiseuille Lab ExperimentDocument7 pagesPoiseuille Lab ExperimentArjun SinghPas encore d'évaluation
- AIX PowerHA (HACMP) CommandsDocument3 pagesAIX PowerHA (HACMP) CommandsdanilaixPas encore d'évaluation
- 88 Numerical Series BASED TECHNOLOGY AWARENESS Grigory GRABOVOIJADocument5 pages88 Numerical Series BASED TECHNOLOGY AWARENESS Grigory GRABOVOIJAStellaEstel93% (15)
- Group 2 (Oacc39)Document3 pagesGroup 2 (Oacc39)viernazanne.viadoPas encore d'évaluation
- Kristian Tlangau - November, 2016 PDFDocument52 pagesKristian Tlangau - November, 2016 PDFMizoram Presbyterian Church SynodPas encore d'évaluation
- InternshipDocument14 pagesInternshipMohammed Shaheeruddin0% (1)
- David Beard Composer CV ShortDocument2 pagesDavid Beard Composer CV ShortEhsan KarimyPas encore d'évaluation
- Econ 103 - 01Document3 pagesEcon 103 - 01perrerPas encore d'évaluation
- Super CatalogueDocument8 pagesSuper CatalogueITL200_UPas encore d'évaluation
- Tesco, PLC "From Mouse To House"Document13 pagesTesco, PLC "From Mouse To House"Md IrfanPas encore d'évaluation
- Different Types of Steering Systems + ExamplesDocument0 pageDifferent Types of Steering Systems + ExamplesAbhilash NagavarapuPas encore d'évaluation
- A Modified Vince Gingery PlasticDocument13 pagesA Modified Vince Gingery PlasticgeppaPas encore d'évaluation
- NNH4-65C-R6-V2: Electrical SpecificationsDocument4 pagesNNH4-65C-R6-V2: Electrical SpecificationsAntony López GálvezPas encore d'évaluation
- Documents - The New Ostpolitik and German-German Relations: Permanent LegationsDocument5 pagesDocuments - The New Ostpolitik and German-German Relations: Permanent LegationsKhairun Nisa JPas encore d'évaluation
- Cooling Tower 3DTrasar ManualDocument90 pagesCooling Tower 3DTrasar ManualArevaLemaPas encore d'évaluation
- Gudenaaparken (Randers) - All You Need To Know BEFORE You GoDocument8 pagesGudenaaparken (Randers) - All You Need To Know BEFORE You GoElaine Zarb GiorgioPas encore d'évaluation








![Risk Management for Medical Device Manufacturers: [MD and IVD]](https://imgv2-1-f.scribdassets.com/img/word_document/602872428/149x198/825d3b5cd7/1666718194?v=1)