Vous aimerez peut-être aussi
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- 12 types of votersDocument13 pages12 types of votersAshok KumarPas encore d'évaluation
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- Fire Safety Awareness - Completion - Certificate PDFDocument1 pageFire Safety Awareness - Completion - Certificate PDFAshok KumarPas encore d'évaluation
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Banking Awareness MCQs For IBPS Clerk Mains 2019 QuestionsDocument12 pagesBanking Awareness MCQs For IBPS Clerk Mains 2019 QuestionsimthegamePas encore d'évaluation
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (894)
- Latest Development in Banking & Finance Sector PDFDocument11 pagesLatest Development in Banking & Finance Sector PDFAshok KumarPas encore d'évaluation
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- HSE - General Awareness - Environmental Management - Completion - Certificate PDFDocument1 pageHSE - General Awareness - Environmental Management - Completion - Certificate PDFAshok KumarPas encore d'évaluation
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- DefaultDocument1 pageDefaultAshok KumarPas encore d'évaluation
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- QuickRide LogcatDocument68 pagesQuickRide LogcatAshok KumarPas encore d'évaluation
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Information Security Awareness - Social Engineering - Completion - CertificateDocument1 pageInformation Security Awareness - Social Engineering - Completion - CertificateAshok KumarPas encore d'évaluation
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- HSE - General Awareness - Environmental Management - Completion - Certificate PDFDocument1 pageHSE - General Awareness - Environmental Management - Completion - Certificate PDFAshok KumarPas encore d'évaluation
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- 20 03 2019 - V2Document41 pages20 03 2019 - V2Ashok KumarPas encore d'évaluation
- Monetary Policy - Strong Dollar Weak DollarDocument24 pagesMonetary Policy - Strong Dollar Weak Dollarmanishkayal100% (1)
- 15,000 New Coronavirus Cases, 786 New Deaths in Past 24 Hours: WHO Report Your ContentDocument9 pages15,000 New Coronavirus Cases, 786 New Deaths in Past 24 Hours: WHO Report Your ContentYour ContentPas encore d'évaluation
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Guide to Atal Pension Yojana benefits for unorganized workersDocument3 pagesGuide to Atal Pension Yojana benefits for unorganized workersds468Pas encore d'évaluation
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- Income Recognition Asset ClassificationDocument5 pagesIncome Recognition Asset ClassificationAshok KumarPas encore d'évaluation
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Pli Ea Bonus PDFDocument2 pagesPli Ea Bonus PDFJayesh Gajare100% (1)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Yugal SurkshaDocument4 pagesYugal Surkshaankit kumarPas encore d'évaluation
- Anticipated Endowment Assurance PDFDocument1 pageAnticipated Endowment Assurance PDFAshok KumarPas encore d'évaluation
- Solo - BASEL III Pillar 3 Disclosures 31-12-2018Document14 pagesSolo - BASEL III Pillar 3 Disclosures 31-12-2018Ashok KumarPas encore d'évaluation
- 1Document3 pages1Ashok KumarPas encore d'évaluation
- APEAMCET2019 CorrectionsDocument1 pageAPEAMCET2019 Correctionsp.narendraPas encore d'évaluation
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Most Important One Liner Questions of October Part-IIDocument11 pagesMost Important One Liner Questions of October Part-IIRobert ShortPas encore d'évaluation
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- 19 03 2019 - V2Document18 pages19 03 2019 - V2Ashok KumarPas encore d'évaluation
- UIIC ClaimFormDocument6 pagesUIIC ClaimFormTeja NagetiPas encore d'évaluation
- 20 03 2019 - V2Document41 pages20 03 2019 - V2Ashok KumarPas encore d'évaluation
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- DefaultDocument1 pageDefaultAshok KumarPas encore d'évaluation
- Insurance Copy.Document4 pagesInsurance Copy.Ashok KumarPas encore d'évaluation
- QuickRide LogcatDocument68 pagesQuickRide LogcatAshok KumarPas encore d'évaluation
- Employee Details Payment & Leave Details: Arrears Current AmountDocument1 pageEmployee Details Payment & Leave Details: Arrears Current AmountAshok KumarPas encore d'évaluation
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- SVR - Constable Prelims KeyDocument6 pagesSVR - Constable Prelims KeyAshok KumarPas encore d'évaluation
- Circle: Bangalore: Circle Head: Shri Ramdas HegdeDocument1 pageCircle: Bangalore: Circle Head: Shri Ramdas HegdeAshok KumarPas encore d'évaluation
- Gen. Math - Quarter 1 Exam (11-HUMS-ALS)Document3 pagesGen. Math - Quarter 1 Exam (11-HUMS-ALS)JerryPas encore d'évaluation
- Online Dispute Resolution For Smart ContractsDocument24 pagesOnline Dispute Resolution For Smart ContractsRidhima SharmaPas encore d'évaluation
- Chapter 3: Computer Instructions: ObjectivesDocument7 pagesChapter 3: Computer Instructions: ObjectivesSteffany RoquePas encore d'évaluation
- HPLC Guide for Pharma Analysis by Oona McPolinDocument3 pagesHPLC Guide for Pharma Analysis by Oona McPolinOmar Cruz Ilustración0% (1)
- Modumate Raises $1.5M Seed Funding To Automate Drafting For ArchitectsDocument2 pagesModumate Raises $1.5M Seed Funding To Automate Drafting For ArchitectsPR.comPas encore d'évaluation
- SSS Format DescriptionDocument5 pagesSSS Format Descriptionmartinos22Pas encore d'évaluation
- Description and Operation: Component Maintenance ManualDocument8 pagesDescription and Operation: Component Maintenance ManualLou Parker100% (1)
- AI QuestionsDocument2 pagesAI QuestionsNarender SinghPas encore d'évaluation
- Audio Encryption Optimization: Harsh Bijlani Dikshant Gupta Mayank LovanshiDocument5 pagesAudio Encryption Optimization: Harsh Bijlani Dikshant Gupta Mayank LovanshiAman Kumar TrivediPas encore d'évaluation
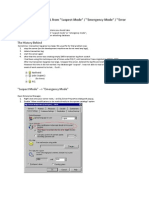
- How To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Document3 pagesHow To Recover MSSQL From Suspect Mode Emergency Mode Error 1813Scott NeubauerPas encore d'évaluation
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- HR Practices and Organizational Strategies in Select: IT Companies in India "Document59 pagesHR Practices and Organizational Strategies in Select: IT Companies in India "Sneha MehtaPas encore d'évaluation
- Advanced Excel Formulas and FunctionsDocument80 pagesAdvanced Excel Formulas and FunctionsSanjay MorePas encore d'évaluation
- Guidline Nurse Education TodayDocument15 pagesGuidline Nurse Education TodayaminPas encore d'évaluation
- Pspice Model For Flicker Noise PDFDocument11 pagesPspice Model For Flicker Noise PDFmikedelta28Pas encore d'évaluation
- Chapter 1 PPT CondensedDocument5 pagesChapter 1 PPT CondensedRaymond GuillartesPas encore d'évaluation
- Integration of Struts, Spring and Hibernate For An University Management SystemDocument8 pagesIntegration of Struts, Spring and Hibernate For An University Management SystemDrajan LamaPas encore d'évaluation
- Commercial Building Maintenance and External Cleaning CompanyDocument8 pagesCommercial Building Maintenance and External Cleaning Companybisankhe2Pas encore d'évaluation
- Tieng Anh 10 Friends Global - Unit 8 - Test 2Document8 pagesTieng Anh 10 Friends Global - Unit 8 - Test 2haminhpham1708Pas encore d'évaluation
- MCQ - Unit 6 AutomationDocument3 pagesMCQ - Unit 6 AutomationDipak naikPas encore d'évaluation
- LIT1126 ACT20M Datasheet 03 14Document16 pagesLIT1126 ACT20M Datasheet 03 14alltheloveintheworldPas encore d'évaluation
- DX5 EuserguideDocument20 pagesDX5 Euserguidegabriel ferreyraPas encore d'évaluation
- Final Year Project Format - UnerDocument5 pagesFinal Year Project Format - UnerDangyi GodSeesPas encore d'évaluation
- AOPEN DEV8430 Preliminary DatasheetDocument2 pagesAOPEN DEV8430 Preliminary DatasheetMarisa García CulpiánPas encore d'évaluation
- Access Token Stealing On WindowsDocument11 pagesAccess Token Stealing On Windows@4e4enPas encore d'évaluation
- Digital Notebook Instruction + HelloDocument9 pagesDigital Notebook Instruction + HelloBelen DozaPas encore d'évaluation
- Visi Misi Ketos 20Document11 pagesVisi Misi Ketos 20haidar alyPas encore d'évaluation
- Winter 2009Document48 pagesWinter 2009ed bookerPas encore d'évaluation
- 115 SstigDocument109 pages115 SstigMahesh DubliPas encore d'évaluation
- June Exam 2018 Feedback.Document3 pagesJune Exam 2018 Feedback.Jordan MorrisPas encore d'évaluation
- MOAC Virtual Lab ExercisesDocument3 pagesMOAC Virtual Lab ExercisesJyoti PatelPas encore d'évaluation
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeD'EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeÉvaluation : 4.5 sur 5 étoiles4.5/5 (3)
- Stuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldD'EverandStuff Matters: Exploring the Marvelous Materials That Shape Our Man-Made WorldÉvaluation : 4 sur 5 étoiles4/5 (289)
- Chemistry for Breakfast: The Amazing Science of Everyday LifeD'EverandChemistry for Breakfast: The Amazing Science of Everyday LifeÉvaluation : 4.5 sur 5 étoiles4.5/5 (14)
- Guidelines for Asset Integrity ManagementD'EverandGuidelines for Asset Integrity ManagementÉvaluation : 5 sur 5 étoiles5/5 (1)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeD'EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction in the Science of Everyday LifeÉvaluation : 4 sur 5 étoiles4/5 (9)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsD'EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsÉvaluation : 4 sur 5 étoiles4/5 (146)
- Science Goes Viral: Captivating Accounts of Science in Everyday LifeD'EverandScience Goes Viral: Captivating Accounts of Science in Everyday LifeÉvaluation : 5 sur 5 étoiles5/5 (1)