Vous aimerez peut-être aussi
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- Vibration Exciter (2N To 100N) : Use ScenariosDocument1 pageVibration Exciter (2N To 100N) : Use ScenariosVignesh SelvarajPas encore d'évaluation
- 1page 3Document2 pages1page 3Sajid MahmoodPas encore d'évaluation
- Pitot Static Tube With Digital ManometerDocument4 pagesPitot Static Tube With Digital ManometerVignesh SelvarajPas encore d'évaluation
- 1200W Smoke Generator:: Technical DetailsDocument1 page1200W Smoke Generator:: Technical DetailsVignesh SelvarajPas encore d'évaluation
- Impact Test MachineDocument2 pagesImpact Test MachineVignesh SelvarajPas encore d'évaluation
- Service DetailsDocument1 pageService DetailsVignesh SelvarajPas encore d'évaluation
- Carbon PDFDocument4 pagesCarbon PDFVignesh SelvarajPas encore d'évaluation
- AA02 Supersonic Wind Tunnel ContinuousDocument5 pagesAA02 Supersonic Wind Tunnel ContinuousVignesh SelvarajPas encore d'évaluation
- Impact Testing Machine Make KedarDocument3 pagesImpact Testing Machine Make KedarVignesh SelvarajPas encore d'évaluation
- Linear Variable Differential TransformerDocument1 pageLinear Variable Differential TransformerVignesh SelvarajPas encore d'évaluation
- Analysis of An Inflatable Gossamer Device To Efficiently De-OrbitDocument100 pagesAnalysis of An Inflatable Gossamer Device To Efficiently De-OrbitVignesh SelvarajPas encore d'évaluation
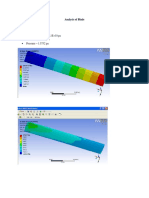
- Analysis of BladeDocument3 pagesAnalysis of BladeVignesh SelvarajPas encore d'évaluation
- EI P L: Column Testing - Fixed-FixedDocument3 pagesEI P L: Column Testing - Fixed-FixedVignesh SelvarajPas encore d'évaluation
- TicDocument1 pageTicVignesh SelvarajPas encore d'évaluation
- Member Management System - MMS User GuideDocument11 pagesMember Management System - MMS User GuideVignesh SelvarajPas encore d'évaluation
- Sample Employment Application FormDocument4 pagesSample Employment Application Formapi-296940651Pas encore d'évaluation
- Newton's RingDocument7 pagesNewton's RingReddyvari Venugopal100% (1)
- Thulasi StotramDocument2 pagesThulasi StotramVignesh SelvarajPas encore d'évaluation
- Quantitative AptitudeDocument2 pagesQuantitative AptitudeSivakumar MPas encore d'évaluation
- Form 1: Employee Personal Information Name of DepartmentDocument12 pagesForm 1: Employee Personal Information Name of DepartmentRajashree PatilPas encore d'évaluation
- Pre CompDocument33 pagesPre CompVignesh SelvarajPas encore d'évaluation
- Achievements Summary-AadesDocument1 pageAchievements Summary-AadesVignesh SelvarajPas encore d'évaluation
- Vinayakar-Subramanyar ThuthiDocument1 pageVinayakar-Subramanyar ThuthiVignesh SelvarajPas encore d'évaluation
- Sample Employment Application FormDocument4 pagesSample Employment Application Formapi-296940651Pas encore d'évaluation
- Vocab Spot CheckDocument9 pagesVocab Spot CheckMr. Yogesh RamaniPas encore d'évaluation
- ShivaDocument6 pagesShivaVignesh SelvarajPas encore d'évaluation
- Optimization Formulations For Composite Structures Subjected To Compression LoadsDocument2 pagesOptimization Formulations For Composite Structures Subjected To Compression LoadsVignesh SelvarajPas encore d'évaluation
- Bottom view scale 1:3 doc 2Document1 pageBottom view scale 1:3 doc 2Vignesh SelvarajPas encore d'évaluation
- Length-2.6 M Chord 0.15m No of Blade-5 Radius-1.2 M Max Torque - 150 N-M Rated Wind Speed-11 M/s Max Force - 500N, Operational Force-250 N NACA-4415Document1 pageLength-2.6 M Chord 0.15m No of Blade-5 Radius-1.2 M Max Torque - 150 N-M Rated Wind Speed-11 M/s Max Force - 500N, Operational Force-250 N NACA-4415Vignesh SelvarajPas encore d'évaluation
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Jithesh Balakrishnan ResumeDocument3 pagesJithesh Balakrishnan Resumejithesh_2Pas encore d'évaluation
- UntitledDocument155 pagesUntitledpriyait182Pas encore d'évaluation
- IBC 2016 TABLE 1604.3 DEFLECTION LIMITSDocument1 pageIBC 2016 TABLE 1604.3 DEFLECTION LIMITSMuhammad Najam AbbasPas encore d'évaluation
- Civil Engineering - Water and Waste Water - Earthquake - RailwaysDocument134 pagesCivil Engineering - Water and Waste Water - Earthquake - RailwaysNavid NakhaiePas encore d'évaluation
- Building PermitDocument2 pagesBuilding PermitRebecca LeysonPas encore d'évaluation
- CCNA Practical Lab 2nd Edition PDFDocument88 pagesCCNA Practical Lab 2nd Edition PDFEl Amrani LailaPas encore d'évaluation
- J.Jesudas: Abu Dhabi, UAE. Mobile:050-4693538 Driving License No: 815678Document9 pagesJ.Jesudas: Abu Dhabi, UAE. Mobile:050-4693538 Driving License No: 815678aniljesudasPas encore d'évaluation
- 03-AHU Schedule 16-02-11Document23 pages03-AHU Schedule 16-02-11karunvandnaPas encore d'évaluation
- Asana: Your Ultimate Task Management ToolDocument113 pagesAsana: Your Ultimate Task Management ToolHeartie Queen Tamayo100% (2)
- Palm IslandDocument4 pagesPalm IslandHemkantYaduvanshiPas encore d'évaluation
- Basics Configuration of PBX Nortel Meridain 81cDocument26 pagesBasics Configuration of PBX Nortel Meridain 81cGil HalePas encore d'évaluation
- Chapter 7 Securing Information Systems AnswersDocument13 pagesChapter 7 Securing Information Systems AnswersTanpopo Ikuta100% (1)
- RCD ObjectivesDocument3 pagesRCD ObjectivesiamcerbzjrPas encore d'évaluation
- History, Answer, Quiz 03Document3 pagesHistory, Answer, Quiz 03JARMRAPas encore d'évaluation
- Intermec Enable Scanner Port Is Grayed Out in Internal Scanner Options On An Intermec Mobile ComputerDocument3 pagesIntermec Enable Scanner Port Is Grayed Out in Internal Scanner Options On An Intermec Mobile Computercocibolca61Pas encore d'évaluation
- Tarek DocumentDocument5 pagesTarek DocumentTarek Mohamad100% (1)
- 5 Min Guide HiPath 3800 - 15 - 07 - 11Document2 pages5 Min Guide HiPath 3800 - 15 - 07 - 11CheickOumarDiabyPas encore d'évaluation
- DIAL Catalogue 2011Document44 pagesDIAL Catalogue 2011Cecilia GonzalezPas encore d'évaluation
- EstimateDocument28 pagesEstimateKuruvaJanardhanPas encore d'évaluation
- Lab 1 StructureDocument11 pagesLab 1 StructureVerra Myza AratPas encore d'évaluation
- Standard Growth RoomsDocument8 pagesStandard Growth Roomsapi-177042586Pas encore d'évaluation
- Inspection and Test Plans (ITP) - MUM-06 Pipework Installation ChecklistDocument3 pagesInspection and Test Plans (ITP) - MUM-06 Pipework Installation Checklistnice guyPas encore d'évaluation
- Architectural Supplemental ServicesDocument60 pagesArchitectural Supplemental ServicesLuwella GumallaoiPas encore d'évaluation
- Amvic ICF Installation ManualDocument136 pagesAmvic ICF Installation ManualdanPas encore d'évaluation
- Chain shackle specifications and applicationsDocument1 pageChain shackle specifications and applicationskariem noweerPas encore d'évaluation
- War 3 PatchDocument9 pagesWar 3 Patchcarlos eduardo palma liceaPas encore d'évaluation
- Reasoning Class AssignmentDocument21 pagesReasoning Class AssignmentBindu SairaPas encore d'évaluation
- Fourth Year Design Brief 2021-22Document4 pagesFourth Year Design Brief 2021-22Sandip SharmaPas encore d'évaluation
- Oracle Nosql Database,: 12.1.3.5, COMMUNITY EDITIONDocument5 pagesOracle Nosql Database,: 12.1.3.5, COMMUNITY EDITIONCarlos Mario HerreraPas encore d'évaluation
- Personal details and career profile of Hembala KashyapDocument2 pagesPersonal details and career profile of Hembala KashyapHembala KashyapPas encore d'évaluation