Vous aimerez peut-être aussi
- Energies 02 00377Document34 pagesEnergies 02 00377MANAS KUMAR RATHPas encore d'évaluation
- Supported Electrolyte Thin Film Synthesis For SofcDocument16 pagesSupported Electrolyte Thin Film Synthesis For SofcMANAS KUMAR RATHPas encore d'évaluation
- Cation-Exchange Induced High Power Electrochemical Properties of Core-ShellDocument7 pagesCation-Exchange Induced High Power Electrochemical Properties of Core-ShellMANAS KUMAR RATHPas encore d'évaluation
- A Mathematical Model of Crystallization in An EmulsionDocument13 pagesA Mathematical Model of Crystallization in An EmulsionMANAS KUMAR RATHPas encore d'évaluation
- Solid-State Re Activity at Hetrophase InterfaceDocument43 pagesSolid-State Re Activity at Hetrophase InterfaceMANAS KUMAR RATHPas encore d'évaluation
- Preparation and Characterization of Size-Controlled CeO2Document5 pagesPreparation and Characterization of Size-Controlled CeO2MANAS KUMAR RATHPas encore d'évaluation
- A Colloidal Approach To Nanometre-Sized Mixed Oxide CeramicDocument6 pagesA Colloidal Approach To Nanometre-Sized Mixed Oxide CeramicMANAS KUMAR RATHPas encore d'évaluation
- Sofc Understanding Materials CompatibilityDocument32 pagesSofc Understanding Materials CompatibilityMANAS KUMAR RATHPas encore d'évaluation
- Interface FractureDocument28 pagesInterface FractureMANAS KUMAR RATHPas encore d'évaluation
- Oxide Ion ElectrolytesDocument40 pagesOxide Ion ElectrolytesMANAS KUMAR RATHPas encore d'évaluation
- Recent Advances in Materials For SofcDocument33 pagesRecent Advances in Materials For SofcMANAS KUMAR RATH100% (3)
- New Electrocatalysts by RialDocument29 pagesNew Electrocatalysts by RialMANAS KUMAR RATHPas encore d'évaluation
- A Review of Anode Materials Development in SolidDocument35 pagesA Review of Anode Materials Development in SolidMANAS KUMAR RATH100% (1)
- Ferroelectric Devices: Kenji UchinoDocument10 pagesFerroelectric Devices: Kenji UchinoMANAS KUMAR RATHPas encore d'évaluation
- Text Chap8Document22 pagesText Chap8MANAS KUMAR RATH100% (1)
- Text Chap6Document14 pagesText Chap6MANAS KUMAR RATHPas encore d'évaluation
- Text Chap9Document12 pagesText Chap9MANAS KUMAR RATH100% (2)
- 11 Future of Ferroelectric DevicesDocument29 pages11 Future of Ferroelectric DevicesMANAS KUMAR RATH100% (1)
- Text Chapt10Document20 pagesText Chapt10MANAS KUMAR RATHPas encore d'évaluation
- Text Chap5Document12 pagesText Chap5MANAS KUMAR RATHPas encore d'évaluation
- Text Chap3Document48 pagesText Chap3MANAS KUMAR RATH100% (1)
- Text Chap4Document14 pagesText Chap4MANAS KUMAR RATHPas encore d'évaluation
- Text Chap2Document34 pagesText Chap2MANAS KUMAR RATHPas encore d'évaluation
- Text Chap1Document22 pagesText Chap1MANAS KUMAR RATHPas encore d'évaluation
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- 不锈钢知识Document20 pages不锈钢知识Mrinal Kanti BhaduriPas encore d'évaluation
- COOKERY 9 - Q1 - Mod2 Week 2 TrueDocument26 pagesCOOKERY 9 - Q1 - Mod2 Week 2 TrueLeah Rizza CabaliwPas encore d'évaluation
- MDS Report Substances of Assemblies and Materials: 1. Company and Product NameDocument3 pagesMDS Report Substances of Assemblies and Materials: 1. Company and Product NameMohamed HarisPas encore d'évaluation
- ArmaFlex FRV Product Brochure ASEAN PrintDocument12 pagesArmaFlex FRV Product Brochure ASEAN PrintMinhSon HaPas encore d'évaluation
- Answers - Mole Concept - VA - Redox Extra WorksheetDocument22 pagesAnswers - Mole Concept - VA - Redox Extra WorksheetSundaravadivel Prabhav (Njc)Pas encore d'évaluation
- Bs en Standard Code List CompressDocument64 pagesBs en Standard Code List CompressJAHANGEER BASHAPas encore d'évaluation
- "The Comparison of Stamicarbon and Saipem Urea Technology": October 2016Document11 pages"The Comparison of Stamicarbon and Saipem Urea Technology": October 2016Anonymous iuFpXLPas encore d'évaluation
- Final Report NDT PDFDocument8 pagesFinal Report NDT PDFAbhay Shankar MishraPas encore d'évaluation
- Superduplex Stainless Steel Article-4Document3 pagesSuperduplex Stainless Steel Article-4mengelito almontePas encore d'évaluation
- Fundamentals of Stripe CoatingDocument164 pagesFundamentals of Stripe Coatinganangwahjudi100% (1)
- Source Workspace Acoustic SolutionsDocument13 pagesSource Workspace Acoustic Solutionslea G Plummer100% (1)
- Colloids and SuspensionsDocument4 pagesColloids and SuspensionsJulius Macaballug100% (1)
- 228-04 KALTEK SG Iron French FoundryDocument4 pages228-04 KALTEK SG Iron French FoundryMar CarreonPas encore d'évaluation
- Novel Process Concept For The Production of H2 and H2SO4 by SO2-Depolarized ElectrolysisDocument12 pagesNovel Process Concept For The Production of H2 and H2SO4 by SO2-Depolarized ElectrolysisFelipe_ArVPas encore d'évaluation
- Mechanical Progress Report SAMPLEDocument14 pagesMechanical Progress Report SAMPLERebecca WattsPas encore d'évaluation
- Rheobuild SP1: A High Range Water Reducing Superplasticising Admixture For The Production of Rheoplastic ConcreteDocument2 pagesRheobuild SP1: A High Range Water Reducing Superplasticising Admixture For The Production of Rheoplastic ConcreteFrancois-Pas encore d'évaluation
- Schunk Carbon Technology Carbon Slide Bearings enDocument11 pagesSchunk Carbon Technology Carbon Slide Bearings enamr yosryPas encore d'évaluation
- V FCCDocument38 pagesV FCCpipe_boyPas encore d'évaluation
- Codex Standard For Edam: CODEX STAN 265-1966 Page 1 of 5Document5 pagesCodex Standard For Edam: CODEX STAN 265-1966 Page 1 of 5hoda hassanPas encore d'évaluation
- Ampcoloy 972: Technical Data SheetDocument1 pageAmpcoloy 972: Technical Data SheetmazaherramazaniPas encore d'évaluation
- Installation of Marble and Granite Tiles On FloorDocument7 pagesInstallation of Marble and Granite Tiles On FloorAleen Gamal Al-Dinji100% (2)
- GLASSDocument7 pagesGLASSGatut SulianaPas encore d'évaluation
- CBSE Class 12 Chemistry Quick Revision Notes Co-Ordination CompoundsDocument8 pagesCBSE Class 12 Chemistry Quick Revision Notes Co-Ordination CompoundsAbid waniPas encore d'évaluation
- Metal Activity Series Virtual LabDocument5 pagesMetal Activity Series Virtual LabFarhan HabibzaiPas encore d'évaluation
- DM Water Flow ChartDocument4 pagesDM Water Flow ChartAshutosh GirdharPas encore d'évaluation
- Chapter 15 (Acid and Bases)Document46 pagesChapter 15 (Acid and Bases)aliefyan4769Pas encore d'évaluation
- ASTM C1708 Self-Leveling Mortars Containing Hydraulic CementsDocument8 pagesASTM C1708 Self-Leveling Mortars Containing Hydraulic CementsIqbal KhanPas encore d'évaluation
- 10.betonske Konstrukcije - Prvi Deo - 8 PDFDocument4 pages10.betonske Konstrukcije - Prvi Deo - 8 PDFIvan JovanovicPas encore d'évaluation
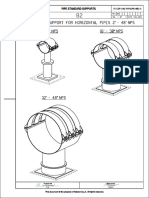
- Clamped Base Support For Horizontal Pipes 2" - 48" NPS: Pipe Standard SupportsDocument2 pagesClamped Base Support For Horizontal Pipes 2" - 48" NPS: Pipe Standard SupportsMainuddin AliPas encore d'évaluation
- Self-Healing at The Cut Edge of Coil-Coated Galvanised SteelDocument145 pagesSelf-Healing at The Cut Edge of Coil-Coated Galvanised SteelEwo50 NewPas encore d'évaluation