Vous aimerez peut-être aussi
- Kuliah MG 9 Racemix Mixture and ResolutionDocument184 pagesKuliah MG 9 Racemix Mixture and ResolutionBowo Aank ApriantoPas encore d'évaluation
- The Determination of Carboxylic Functional Groups: Monographs in Organic Functional Group AnalysisD'EverandThe Determination of Carboxylic Functional Groups: Monographs in Organic Functional Group AnalysisPas encore d'évaluation
- Problems On Named ReactionsDocument103 pagesProblems On Named ReactionsBapu ThoratPas encore d'évaluation
- PMR Spectroscopy: Solved Problems Volume : IID'EverandPMR Spectroscopy: Solved Problems Volume : IIÉvaluation : 5 sur 5 étoiles5/5 (3)
- Chapter 5 Elimination Reaction - 2016Document19 pagesChapter 5 Elimination Reaction - 2016Syuhadah NoordinPas encore d'évaluation
- Aromaticity 2019Document65 pagesAromaticity 2019Shreya PrakashPas encore d'évaluation
- Stereochemistry by Qari Abdullah SiddiqueDocument48 pagesStereochemistry by Qari Abdullah SiddiqueABDULLAH0% (1)
- Aromaticity CompleteDocument104 pagesAromaticity Completewahidalwahdi100% (1)
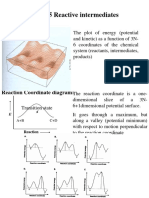
- Chap. 5 Reactive Intermediates: Energy SurfaceDocument20 pagesChap. 5 Reactive Intermediates: Energy SurfaceAnil KumarPas encore d'évaluation
- Org Chem Sem 3 Paper 2Document15 pagesOrg Chem Sem 3 Paper 2Rohit DeshmukhPas encore d'évaluation
- Practice Problems On Addition Reactions To Alkenes With AnswersDocument4 pagesPractice Problems On Addition Reactions To Alkenes With AnswersSangetha ChelladoraiPas encore d'évaluation
- Resonance and Inductive Effects PresentationDocument36 pagesResonance and Inductive Effects Presentationeagl33yePas encore d'évaluation
- QOI0809 AlkenesDocument30 pagesQOI0809 Alkenesmtucker17Pas encore d'évaluation
- Sharpless Asymmetric EpoxidationDocument19 pagesSharpless Asymmetric Epoxidationjaanabhenchod100% (2)
- Atropisomerism PDFDocument22 pagesAtropisomerism PDFAnil KumarPas encore d'évaluation
- Fused - Heterocycles NomenclatureDocument26 pagesFused - Heterocycles Nomenclaturelawanya pandeyPas encore d'évaluation
- Reactions of Alcohols: Organic Chemistry, 7Document53 pagesReactions of Alcohols: Organic Chemistry, 7haha_le12100% (1)
- Organic Chemistry Midterm 1 Dir+eff++keyDocument1 pageOrganic Chemistry Midterm 1 Dir+eff++keyNorma Leticia RamosPas encore d'évaluation
- 04 Reactive IntermediatesDocument115 pages04 Reactive IntermediatesMuhammad ArsalanPas encore d'évaluation
- TerpenoidsDocument17 pagesTerpenoidsSayli Sawant100% (1)
- Pericyclics-2014 Handout PDFDocument79 pagesPericyclics-2014 Handout PDFnavchemPas encore d'évaluation
- Neighbouring Group Participation or NGP inDocument4 pagesNeighbouring Group Participation or NGP inbharatbhushansankhya100% (1)
- Iub Pha404 Autumn 2022 Ms BasicDocument52 pagesIub Pha404 Autumn 2022 Ms BasicTanvir FahimPas encore d'évaluation
- Lab ManualDocument19 pagesLab Manualanon_467104036Pas encore d'évaluation
- Organic Chemistry, Second Edition Janice Gorzynski Smith, ch1Document40 pagesOrganic Chemistry, Second Edition Janice Gorzynski Smith, ch1sungyeon heoPas encore d'évaluation
- Sni, Nighbouring GP Participation & E1cbDocument19 pagesSni, Nighbouring GP Participation & E1cbsaheedvkPas encore d'évaluation
- 01 StereochemistryDocument6 pages01 StereochemistryGundum Bodyz100% (1)
- Heteocyclic CompoundsDocument116 pagesHeteocyclic CompoundsLetin Shrivastav100% (1)
- Molecular OrbitalsDocument80 pagesMolecular Orbitals1balamanian100% (1)
- 01.coordination Chemistry Class Notes Part I-1 PDFDocument86 pages01.coordination Chemistry Class Notes Part I-1 PDFShadrack Peter100% (1)
- Symmetry Operations and Point GroupDocument13 pagesSymmetry Operations and Point GroupRahul AroraPas encore d'évaluation
- Protection Groups in Organic PDFDocument67 pagesProtection Groups in Organic PDFToàn MinhPas encore d'évaluation
- Bio-Inorganic ChemistryDocument66 pagesBio-Inorganic ChemistryPhalynxPas encore d'évaluation
- Chemistry of Natural Products: (CHEM-479)Document13 pagesChemistry of Natural Products: (CHEM-479)Sidra Tul MuntahaPas encore d'évaluation
- PolimerDocument22 pagesPolimerDhea Kana ZhafiraPas encore d'évaluation
- Huckel Theory For Conjugated Systems: CH 105: Organic ChemistryDocument72 pagesHuckel Theory For Conjugated Systems: CH 105: Organic ChemistryRaunaq Bhirangi100% (1)
- Inductive EffectDocument38 pagesInductive EffectJoe JPas encore d'évaluation
- Ugi ReactionDocument11 pagesUgi ReactionNavnath HatvatePas encore d'évaluation
- Hückel's MO Treatment of BenzeneDocument12 pagesHückel's MO Treatment of BenzeneRichard Allen0% (1)
- Organometallic CompoundsDocument66 pagesOrganometallic CompoundsJon Ho100% (1)
- Nomenclature Sheet 2021,13thDocument89 pagesNomenclature Sheet 2021,13thsane jha vlogsPas encore d'évaluation
- Manual de Toxicologia (Ingles)Document390 pagesManual de Toxicologia (Ingles)Raúl Israel Ccorimanya PalominoPas encore d'évaluation
- SCH 302 Axial ChiralityDocument22 pagesSCH 302 Axial ChiralityArangaPas encore d'évaluation
- Organic Chemistry II Practice Exam #3A Answer KeyDocument8 pagesOrganic Chemistry II Practice Exam #3A Answer Keyhiep237Pas encore d'évaluation
- Csir Model QuestionsDocument15 pagesCsir Model QuestionsbaluPas encore d'évaluation
- HybridizationDocument18 pagesHybridizationSoub kuopPas encore d'évaluation
- Retrosynthetic Analysis PDFDocument6 pagesRetrosynthetic Analysis PDFNolePas encore d'évaluation
- E1 and E2 ReactionsDocument30 pagesE1 and E2 ReactionsVidhu Pandey100% (1)
- Neighbouring Group ParticipationDocument15 pagesNeighbouring Group ParticipationAbdulMananPas encore d'évaluation
- CH2203 - Spectroscopy of Inorganic CompoundsDocument6 pagesCH2203 - Spectroscopy of Inorganic CompoundsJohnPas encore d'évaluation
- C - C Bond Formation PDFDocument14 pagesC - C Bond Formation PDFZee_ShaniPas encore d'évaluation
- Experiment 1: Preparation of 2-Iodobenzoic Acid From Anthranilic Acid (2-Amino Benzoic Acid)Document11 pagesExperiment 1: Preparation of 2-Iodobenzoic Acid From Anthranilic Acid (2-Amino Benzoic Acid)Sanjida Khandoker 1911009049Pas encore d'évaluation
- Chapter 05 Wade 8thDocument66 pagesChapter 05 Wade 8thanupamgupta112Pas encore d'évaluation
- Aldehydes and KetonesDocument13 pagesAldehydes and Ketonesaleah kimPas encore d'évaluation
- Structure and Synthesis of Alcohols: Organic Chemistry, 7Document52 pagesStructure and Synthesis of Alcohols: Organic Chemistry, 7haha_le12100% (1)
- Alkyl Halides and Nucleophilic SubstitutionDocument53 pagesAlkyl Halides and Nucleophilic SubstitutionRaja DanishPas encore d'évaluation
- Reaction Guide by James Ashenhurst. 1-James AshenhurstDocument76 pagesReaction Guide by James Ashenhurst. 1-James AshenhurstSankar AdhikariPas encore d'évaluation
- Chapter - 5 Coordination CompoundsDocument11 pagesChapter - 5 Coordination CompoundsSuresh Dasaraddi67% (3)
- Chemical Bonding and Molecular StructureDocument10 pagesChemical Bonding and Molecular Structuresuper royPas encore d'évaluation
- Atomic StructureDocument8 pagesAtomic StructureGagan NdPas encore d'évaluation
- AllylPO 1Document20 pagesAllylPO 1evsgoud_goudPas encore d'évaluation
- Diphenyl Allylamine PODocument8 pagesDiphenyl Allylamine POevsgoud_goudPas encore d'évaluation
- ThermodynamicsDocument6 pagesThermodynamicsevsgoud_goudPas encore d'évaluation
- StoichiometryDocument8 pagesStoichiometryevsgoud_goudPas encore d'évaluation
- Ally LpoDocument8 pagesAlly Lpoevsgoud_goudPas encore d'évaluation
- AIBN Used DendrimerDocument2 pagesAIBN Used Dendrimerevsgoud_goudPas encore d'évaluation
- APPSC UrlsDocument1 pageAPPSC Urlsevsgoud_goudPas encore d'évaluation
- Structure and Content of CtetDocument3 pagesStructure and Content of Ctetevsgoud_goudPas encore d'évaluation
- Synthesis and Characterization of Some Novel Dendritic Architectures Bearing Chalcone at The PeDocument12 pagesSynthesis and Characterization of Some Novel Dendritic Architectures Bearing Chalcone at The Peevsgoud_goudPas encore d'évaluation
- Synthetic Strategies in ChemistryDocument361 pagesSynthetic Strategies in Chemistryevsgoud_goudPas encore d'évaluation
- APPSC UrlsDocument1 pageAPPSC Urlsevsgoud_goudPas encore d'évaluation
- PE RAM Safety Guide PDFDocument52 pagesPE RAM Safety Guide PDFevsgoud_goudPas encore d'évaluation
- Ruthenium Paper PDFDocument6 pagesRuthenium Paper PDFevsgoud_goudPas encore d'évaluation
- Syllabus PDFDocument9 pagesSyllabus PDFevsgoud_goudPas encore d'évaluation
- Synthesis of (+) - Camptothecin: Organic Chemistry: Technology: National Chemical Laboratory, Pune 411008, India E-MailDocument5 pagesSynthesis of (+) - Camptothecin: Organic Chemistry: Technology: National Chemical Laboratory, Pune 411008, India E-Mailevsgoud_goudPas encore d'évaluation
- Marine Drugs: Antitumor Compounds From Marine ActinomycetesDocument39 pagesMarine Drugs: Antitumor Compounds From Marine Actinomycetesevsgoud_goudPas encore d'évaluation
- Homogeneous Catalysis PDFDocument99 pagesHomogeneous Catalysis PDFevsgoud_goudPas encore d'évaluation
- Inorganic Lab ManualDocument17 pagesInorganic Lab ManualEvs GoudPas encore d'évaluation
- Document3 PDFDocument1 pageDocument3 PDFevsgoud_goudPas encore d'évaluation
- Special Features of Organoboron Chemistry: Boron Is Electrophilic Because of Its Empty P OrbitalDocument15 pagesSpecial Features of Organoboron Chemistry: Boron Is Electrophilic Because of Its Empty P Orbitalevsgoud_goud100% (2)
- Hypervalent Iodine: Dess-Martin Periodane: Selective Oxidation of Prim. Alcohols To Aldehydes, Sec. Alcohols To KetonesDocument15 pagesHypervalent Iodine: Dess-Martin Periodane: Selective Oxidation of Prim. Alcohols To Aldehydes, Sec. Alcohols To Ketonesevsgoud_goudPas encore d'évaluation
- Name ReactionsDocument10 pagesName Reactionsevsgoud_goudPas encore d'évaluation
- Solvent - Wikipedia, The Free EncyclopediaDocument13 pagesSolvent - Wikipedia, The Free EncyclopediaEvs GoudPas encore d'évaluation
- Shift Reagents PDFDocument2 pagesShift Reagents PDFevsgoud_goud100% (1)
- CBSE XII Board Paper - Chemistry Set 3 PDFDocument21 pagesCBSE XII Board Paper - Chemistry Set 3 PDFSamkit SharmaPas encore d'évaluation
- Group Meeting - Silicon and FuransDocument19 pagesGroup Meeting - Silicon and Furansevsgoud_goudPas encore d'évaluation
- Physics (Theory) Class XiDocument6 pagesPhysics (Theory) Class XisudeepjosephPas encore d'évaluation
- Iconnect v3.1 User ManualDocument105 pagesIconnect v3.1 User Manualcjamiz2006Pas encore d'évaluation
- 6.Hartl-Clark-Principles of Population GeneticsDocument6 pages6.Hartl-Clark-Principles of Population GeneticsTeflon SlimPas encore d'évaluation
- An Introduction To Computer ArchitectureDocument59 pagesAn Introduction To Computer Architecturekhaled mahmudPas encore d'évaluation
- Spray-Agglomeration of NPK-fertilizer in A Rotating Drum Granulator PDFDocument8 pagesSpray-Agglomeration of NPK-fertilizer in A Rotating Drum Granulator PDFKhoa TrầnPas encore d'évaluation
- Matlab Manual1Document25 pagesMatlab Manual1Richard BrooksPas encore d'évaluation
- Manual GHH Rand CE and CF SeriesDocument58 pagesManual GHH Rand CE and CF SeriesAnonymous 6VCG1YRd50% (4)
- Video Display DevicesDocument66 pagesVideo Display DevicesshabanaPas encore d'évaluation
- Case 1Document2 pagesCase 1Chiks JpegPas encore d'évaluation
- Ib8091en PDFDocument1 pageIb8091en PDFnicu secosanPas encore d'évaluation
- GCSE Higher Student Book Unit Test AnswersDocument26 pagesGCSE Higher Student Book Unit Test Answersswiftmessi100% (5)
- More Than One Answer Is CorrectDocument182 pagesMore Than One Answer Is CorrectNikhil GandhiPas encore d'évaluation
- Chemistry Leuco DyesDocument312 pagesChemistry Leuco Dyeskarel470% (1)
- Machine Design-II Question BankDocument9 pagesMachine Design-II Question BankProf. Avinash MahalePas encore d'évaluation
- Audi 1.8l and 2.0l TFSI Engines of The EA888 Model Family (Third Generation)Document64 pagesAudi 1.8l and 2.0l TFSI Engines of The EA888 Model Family (Third Generation)NTN-Nguyễn Trọng Nghĩa100% (1)
- Annexure 1 - Technical Bid Analysis For Emergency Relief ValveDocument1 pageAnnexure 1 - Technical Bid Analysis For Emergency Relief ValveNikhil KarkeraPas encore d'évaluation
- Catalogo Tarifa Hisense 2016Document37 pagesCatalogo Tarifa Hisense 2016David GarciaPas encore d'évaluation
- IOS Developer - ExperiencedDocument37 pagesIOS Developer - Experiencedswornavidhya.mahadevanPas encore d'évaluation
- Advantage of Oil CoolingDocument2 pagesAdvantage of Oil CoolingYin ThoPas encore d'évaluation
- PumpsDocument8 pagesPumpskannagi198Pas encore d'évaluation
- AndroidDocument61 pagesAndroidNamithaPas encore d'évaluation
- MODULE 0 CPE105 Review On StatisticsDocument12 pagesMODULE 0 CPE105 Review On StatisticsKishiane Ysabelle L. CabaticPas encore d'évaluation
- Wear and Corrosion Properties of AISI 420 Martensitic Stainless Steel Treated by Active Screen Plasma Nitriding PDFDocument9 pagesWear and Corrosion Properties of AISI 420 Martensitic Stainless Steel Treated by Active Screen Plasma Nitriding PDFsaltbathPas encore d'évaluation
- M and V-Issues-and-Examples 2019 - 02 - 04 - FINAL PDFDocument67 pagesM and V-Issues-and-Examples 2019 - 02 - 04 - FINAL PDFNeeraj Singhvi100% (2)
- Pronouns - Maurice PDFDocument2 pagesPronouns - Maurice PDFSCRIBDBUUPas encore d'évaluation
- KEIYU NDT Ultrasonic TransducerDocument6 pagesKEIYU NDT Ultrasonic TransducersrgokuPas encore d'évaluation
- Mid Year Test: Objective QuestionsDocument3 pagesMid Year Test: Objective QuestionsNur SuHaPas encore d'évaluation
- SQL Interview QuestionsDocument2 pagesSQL Interview QuestionscourpediaPas encore d'évaluation
- CD30LA0011EDocument357 pagesCD30LA0011EMichael Naím Dévora QuintanarPas encore d'évaluation
- NC, CNC and RoboticsDocument100 pagesNC, CNC and RoboticsGovt Job update - JagadishPas encore d'évaluation
- ISCSI OptimizationDocument28 pagesISCSI OptimizationrahulhclPas encore d'évaluation