Vous aimerez peut-être aussi
- Urinary Organic Acids: Clinical & Interpretive GuideDocument12 pagesUrinary Organic Acids: Clinical & Interpretive GuidepatelsspPas encore d'évaluation
- Pharmacogenomic and Pharmacogenetics - DIAN PDFDocument34 pagesPharmacogenomic and Pharmacogenetics - DIAN PDFfakultasfarmasiPas encore d'évaluation
- (Journal of Pediatric Endocrinology and Metabolism) Veganism As A Cause of Iodine Deficient HypothyroidismDocument4 pages(Journal of Pediatric Endocrinology and Metabolism) Veganism As A Cause of Iodine Deficient HypothyroidismFuDanielePas encore d'évaluation
- Globocan 2020Document2 pagesGlobocan 2020Dicky keyPas encore d'évaluation
- Psych Case StudyDocument14 pagesPsych Case Studyapi-604581864Pas encore d'évaluation
- Nclex PointersDocument4 pagesNclex PointersCarl Michael de Guzman75% (12)
- Mitochondrial Disorders Overview - GeneReviews® - NCBI Bookshelf HTTPSWWW - Ncbi.nlm - nih.govbooksNBK1224Document24 pagesMitochondrial Disorders Overview - GeneReviews® - NCBI Bookshelf HTTPSWWW - Ncbi.nlm - nih.govbooksNBK1224denisaPas encore d'évaluation
- The Spectrum of Mitochondrial Disease Ep-3-10Document8 pagesThe Spectrum of Mitochondrial Disease Ep-3-10F4AR100% (1)
- Function of MitochondriaDocument5 pagesFunction of MitochondriaArshia NazirPas encore d'évaluation
- Mitochondrial DiseaseDocument12 pagesMitochondrial DiseaseWSETPas encore d'évaluation
- MCQ MembranesDocument10 pagesMCQ MembranesMarilyne RizkPas encore d'évaluation
- COURSE WORK MOLECULAR BIOLOGY & GeneticsDocument3 pagesCOURSE WORK MOLECULAR BIOLOGY & Geneticsusaeed00000Pas encore d'évaluation
- Mitochondrial DiseasesDocument24 pagesMitochondrial DiseasesRugas PribawaPas encore d'évaluation
- Presentation Gut Brain August 2018Document17 pagesPresentation Gut Brain August 2018Sribharathi Indrajith100% (1)
- HMP Pathway PDFDocument44 pagesHMP Pathway PDFHimanshu SharmaPas encore d'évaluation
- The Investigation of Mitochondrial DNA Maintenance Abnormalities in Adult Onset Mitochondrial DiseaseDocument11 pagesThe Investigation of Mitochondrial DNA Maintenance Abnormalities in Adult Onset Mitochondrial DiseaseZarfishan ShabbirPas encore d'évaluation
- Apoptosis Tutorial NotesDocument8 pagesApoptosis Tutorial NotesismealPas encore d'évaluation
- Mitochondria: Dr. Raheela Jabeen Assistant Professor Department of Biochemistry & Biotechnology, WUMDocument30 pagesMitochondria: Dr. Raheela Jabeen Assistant Professor Department of Biochemistry & Biotechnology, WUMMah Noor100% (1)
- What Are CarbohydratesDocument36 pagesWhat Are CarbohydratesPINNACLE CAFEPas encore d'évaluation
- Multidimensional Analysis of Clinical Symptoms in Patients With Fabry's DiseaseDocument12 pagesMultidimensional Analysis of Clinical Symptoms in Patients With Fabry's DiseaseOnon EssayedPas encore d'évaluation
- Oxidative Phosphorylation 1Document22 pagesOxidative Phosphorylation 1fatin nadia100% (1)
- MelasDocument9 pagesMelasmrabhilekhPas encore d'évaluation
- Steroidal Hormones (Testosterone, Progesterone, Estrogen PDFDocument23 pagesSteroidal Hormones (Testosterone, Progesterone, Estrogen PDFJazab ChohanPas encore d'évaluation
- EnzymeDocument16 pagesEnzymeapi-3798760100% (2)
- Introduction To MetabolismDocument27 pagesIntroduction To Metabolismcream oPas encore d'évaluation
- Mitochondrial Diseasees Lancet 2012Document10 pagesMitochondrial Diseasees Lancet 2012Caro SuárezPas encore d'évaluation
- Problem Solving Tests in MCB (Full)Document66 pagesProblem Solving Tests in MCB (Full)Anonymous 2UPF2xPas encore d'évaluation
- Microbial Activities and Intestinal Homeostasis A Delicate Balance Between Health and DiseaseDocument13 pagesMicrobial Activities and Intestinal Homeostasis A Delicate Balance Between Health and DiseaseitomoralesPas encore d'évaluation
- Biochemistry LipidsDocument62 pagesBiochemistry LipidsDianne Joy100% (1)
- Erythropoiesis Lect 3Document18 pagesErythropoiesis Lect 3Pramesh PaudelPas encore d'évaluation
- T-Cell Development in The ThymusDocument1 pageT-Cell Development in The Thymusfysi mackPas encore d'évaluation
- Neurotransmitters and Intro To NeuropeptidesDocument566 pagesNeurotransmitters and Intro To NeuropeptidesJAH100% (3)
- PathophysExam1 Review Course HeroDocument7 pagesPathophysExam1 Review Course HeroTammie Gore100% (1)
- Intracellular SignallingDocument45 pagesIntracellular Signallingsana iqbalPas encore d'évaluation
- Matabolic PathwaysDocument11 pagesMatabolic PathwaysLevi100% (2)
- Endocrine Regulation of Glucose MetabolismDocument15 pagesEndocrine Regulation of Glucose MetabolismSoji PhilipPas encore d'évaluation
- Lipid Profile: Dr. MD Razib HasanDocument26 pagesLipid Profile: Dr. MD Razib HasanRazib HasanPas encore d'évaluation
- Reference Ranges For Blood TestsDocument38 pagesReference Ranges For Blood TestscatalinPas encore d'évaluation
- Glycoproteins and Proteoglycans TTVDocument34 pagesGlycoproteins and Proteoglycans TTVcraigPas encore d'évaluation
- Metabolic Concepts (Lecture 1-3)Document8 pagesMetabolic Concepts (Lecture 1-3)atifzea100% (1)
- 1 - XenobioticsDocument24 pages1 - XenobioticsgeenaksamuelPas encore d'évaluation
- Channels, Carriers, and Pumps: An Introduction to Membrane TransportD'EverandChannels, Carriers, and Pumps: An Introduction to Membrane TransportPas encore d'évaluation
- Respiratory Chain & Oxidative PhosphorylationDocument57 pagesRespiratory Chain & Oxidative PhosphorylationHanifa AffianiPas encore d'évaluation
- Lecture 1Document30 pagesLecture 1حموده ابراهيم يونسPas encore d'évaluation
- Unit 4 Cell CommunicationDocument74 pagesUnit 4 Cell CommunicationBerryPas encore d'évaluation
- Diagnositc Testing: By: Jervy P. Beranrdino, RN, MSNDocument34 pagesDiagnositc Testing: By: Jervy P. Beranrdino, RN, MSNadni_wgPas encore d'évaluation
- Modulator: Dr. P.B.Desai HOD, Dept of Biochemistry. Presenter: DR Vijayetha S. KagwadDocument68 pagesModulator: Dr. P.B.Desai HOD, Dept of Biochemistry. Presenter: DR Vijayetha S. KagwadvijayethaPas encore d'évaluation
- Glucose, Part1Document33 pagesGlucose, Part1SarahPas encore d'évaluation
- PROTEIN METABOLISM Dea Farha Fira Darson FineDocument44 pagesPROTEIN METABOLISM Dea Farha Fira Darson FineFarhati MardhiyahPas encore d'évaluation
- Mitochondrial Dysfunction: Methods in Toxicology, Vol. 2D'EverandMitochondrial Dysfunction: Methods in Toxicology, Vol. 2Lawrence H. LashPas encore d'évaluation
- Biochemistry of Free Radicals and AntioxidantsDocument9 pagesBiochemistry of Free Radicals and Antioxidantsagung ari chandraPas encore d'évaluation
- Mitochondria & Ageing: Extracted From The Presentation by Lee Know, NDDocument4 pagesMitochondria & Ageing: Extracted From The Presentation by Lee Know, NDDavid100% (1)
- Format of Animal Development Practical Report:: Text Book Berbahasa Indonesia Atau Bahasa InggrisDocument14 pagesFormat of Animal Development Practical Report:: Text Book Berbahasa Indonesia Atau Bahasa InggrisSafira Dwi OktavianiPas encore d'évaluation
- Effects of Zinc Supplementation On Serum Lipids A Systematic Review and Meta-AnalysisDocument16 pagesEffects of Zinc Supplementation On Serum Lipids A Systematic Review and Meta-Analysispronto4mePas encore d'évaluation
- Montina L TabletDocument10 pagesMontina L TabletKrish Yemkay100% (1)
- The Role of Diet in Multiple Sclerosis - A ReviewDocument16 pagesThe Role of Diet in Multiple Sclerosis - A ReviewHabib G. Moutran BarrosoPas encore d'évaluation
- 8 Coenzymes and Vitamins (كيمياء حيوية صيدلانية (1 PDFDocument74 pages8 Coenzymes and Vitamins (كيمياء حيوية صيدلانية (1 PDFmaher100% (1)
- Antisense Therapy 2020Document18 pagesAntisense Therapy 2020Calvin HansPas encore d'évaluation
- Antigen Processing Process and MHC AssociationDocument3 pagesAntigen Processing Process and MHC AssociationMahathir Mohmed100% (2)
- Dna Mutation & Repair MechanismDocument23 pagesDna Mutation & Repair MechanismOsama Bin RizwanPas encore d'évaluation
- Gene Transfer in BacteriaDocument46 pagesGene Transfer in BacteriaRenz L. SalumbrePas encore d'évaluation
- Mitochondria: PratibhapariharDocument38 pagesMitochondria: PratibhapariharPatel BansariPas encore d'évaluation
- Family History CollectionDocument1 pageFamily History CollectionNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Session Four: HomeworkDocument1 pageSession Four: HomeworkNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Genomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationDocument7 pagesGenomics For The Child Neurologist Facilitator Guide Session Four Results Intepretation and ApplicationNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Alpers-Huttenlocher FactsheetDocument2 pagesAlpers-Huttenlocher FactsheetNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Results Interpretation & Application Toolkit: ManagementDocument1 pageResults Interpretation & Application Toolkit: ManagementNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Informed Consent Talking PointsDocument4 pagesInformed Consent Talking PointsNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Informed Consent ChecklistDocument2 pagesInformed Consent ChecklistNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Dravet Syndrome FactsheetDocument1 pageDravet Syndrome FactsheetNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Genetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedDocument1 pageGenetic Testing Process Homework: Published June 2013 © Nchpeg All Rights ReservedNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Family History CollectionDocument1 pageFamily History CollectionNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- CMA Provider ResourcesDocument1 pageCMA Provider ResourcesNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- HBOC Red Flags ChecklistDocument1 pageHBOC Red Flags ChecklistNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- HBOC Family History Collection ToolDocument4 pagesHBOC Family History Collection ToolNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
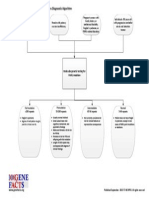
- Fragile X Syndrome Diagnostic AlgorithmDocument1 pageFragile X Syndrome Diagnostic AlgorithmNational Coalition for Health Professional Education in GeneticsPas encore d'évaluation
- Knee, Shin Splints, Ankle Strain, Achilles Tendinitis, Arch Pain, and Many More. All of TheDocument2 pagesKnee, Shin Splints, Ankle Strain, Achilles Tendinitis, Arch Pain, and Many More. All of TheDarko ĐorđevićPas encore d'évaluation
- Endometritis: R.N. Zainab Neamat JumaahDocument19 pagesEndometritis: R.N. Zainab Neamat JumaahKevin Adrian WijayaPas encore d'évaluation
- Apathy Associated With Antidepressant Drugs A Systematic ReviewDocument16 pagesApathy Associated With Antidepressant Drugs A Systematic ReviewJ MrPas encore d'évaluation
- Felocell Felv Helps Prevent Disease Cause by Feline Leukemia Virus (Felv)Document1 pageFelocell Felv Helps Prevent Disease Cause by Feline Leukemia Virus (Felv)Reginaldo ParcianelloPas encore d'évaluation
- Antiviral AgentsDocument14 pagesAntiviral Agentsalishba100% (1)
- Definition:: Pleural EffusionDocument4 pagesDefinition:: Pleural EffusionGetom NgukirPas encore d'évaluation
- Lyme DisorderLLDocument14 pagesLyme DisorderLLLydia Lopz MsnrncdPas encore d'évaluation
- Open Book Exam - Renal Dz. 2019Document2 pagesOpen Book Exam - Renal Dz. 2019Ahmed AliPas encore d'évaluation
- Nasal Polyps Grade IIIDocument2 pagesNasal Polyps Grade IIIRyan John Bito-onPas encore d'évaluation
- Case ManagementDocument11 pagesCase ManagementGabrielle CatalanPas encore d'évaluation
- SMART-COP Score For Pneumonia Severity - MDCalc 2Document1 pageSMART-COP Score For Pneumonia Severity - MDCalc 2johnlau90Pas encore d'évaluation
- SFH - Module 4Document7 pagesSFH - Module 4sammysamuel54321Pas encore d'évaluation
- Guide To Treatment Decision Making For Cleft Type Speech PDFDocument1 pageGuide To Treatment Decision Making For Cleft Type Speech PDFMacarena Paz ÁlvarezPas encore d'évaluation
- Examination of A Male For Determination of PotencyDocument4 pagesExamination of A Male For Determination of PotencyHarish AgarwalPas encore d'évaluation
- Arab Board Exam 2008Document7 pagesArab Board Exam 2008alaaPas encore d'évaluation
- Absent End-Diastolic Velocity in The Umbilical Artery and Its Clinical SignificanceDocument12 pagesAbsent End-Diastolic Velocity in The Umbilical Artery and Its Clinical Significancejakob_saleanPas encore d'évaluation
- Blood Tests & Normal RangeDocument40 pagesBlood Tests & Normal RangeSuria KumarPas encore d'évaluation
- Types of DysrhythmiasDocument15 pagesTypes of DysrhythmiasKevin VillarantePas encore d'évaluation
- Karakteristik Klinis Pasien Blefaroptosis Yang Telah Dilakukan Operasi Di Rumah Sakit Mata Cicendo - Mareta Gustia NingsihDocument8 pagesKarakteristik Klinis Pasien Blefaroptosis Yang Telah Dilakukan Operasi Di Rumah Sakit Mata Cicendo - Mareta Gustia NingsihkarinarakhmaPas encore d'évaluation
- Psycho Pharmacology: by Dr. Alit Aryani SPKJDocument28 pagesPsycho Pharmacology: by Dr. Alit Aryani SPKJMienaPas encore d'évaluation
- Luaran Perdarahan Intraventrikel Yang Dilakukan Operasi Di Departemen Bedah Saraf Rsupn Dr. Cipto MangunkusumoDocument5 pagesLuaran Perdarahan Intraventrikel Yang Dilakukan Operasi Di Departemen Bedah Saraf Rsupn Dr. Cipto MangunkusumoMerry AndrianyPas encore d'évaluation
- Liver Trauma CaseDocument7 pagesLiver Trauma CaseMario KopljarPas encore d'évaluation
- Log Book: Medical Surgical Nursing DepartmentDocument17 pagesLog Book: Medical Surgical Nursing DepartmentMajied MohamedPas encore d'évaluation
- Infective Endocarditis (IE)Document76 pagesInfective Endocarditis (IE)Mahesh RathnayakePas encore d'évaluation
- Dokumen - Tips Loa LoaDocument28 pagesDokumen - Tips Loa LoaSababil AliPas encore d'évaluation
- Psychiatric Nursing: What Is Mental Health Disorder? SilenceDocument11 pagesPsychiatric Nursing: What Is Mental Health Disorder? SilenceJek Dela CruzPas encore d'évaluation
- 23 Cervix Uteri Fact Sheet PDFDocument2 pages23 Cervix Uteri Fact Sheet PDFIrina DeaconescuPas encore d'évaluation