Vous aimerez peut-être aussi
- cp3 TrussdesignDocument106 pagescp3 Trussdesignznyaphotmail.comPas encore d'évaluation
- PCP SynthesisDocument4 pagesPCP SynthesisArturo Burgos100% (1)
- Hs 342Document45 pagesHs 342Fernando Martinez ContrerasPas encore d'évaluation
- Gabapentin hydrochloride process under 38 charsDocument6 pagesGabapentin hydrochloride process under 38 chars13201940100% (1)
- Iodide-Catalyzed Reductions: Development of A Synthesis of Phenylacetic AcidsDocument6 pagesIodide-Catalyzed Reductions: Development of A Synthesis of Phenylacetic AcidsMike Roller100% (1)
- Grade 6 Elevate Science Workbook - 86500313 - 225 - 2022519533Document63 pagesGrade 6 Elevate Science Workbook - 86500313 - 225 - 2022519533satbooks30Pas encore d'évaluation
- Injection Molding SOP for Mini-Jector #55.1 MachineDocument12 pagesInjection Molding SOP for Mini-Jector #55.1 MachineYusuf SethPas encore d'évaluation
- IEEE-Std-C57-149-IEEE Guide For The Application and Interpretation of Frequency Response Analysis For Oil-Immersed Transformers PDFDocument72 pagesIEEE-Std-C57-149-IEEE Guide For The Application and Interpretation of Frequency Response Analysis For Oil-Immersed Transformers PDFJose Luis BarretoPas encore d'évaluation
- Manual 3322006Document46 pagesManual 3322006mallikapathakPas encore d'évaluation
- Gas Treating Technologies and ServicesDocument16 pagesGas Treating Technologies and Services13201940Pas encore d'évaluation
- JIS G 3106 - NewDocument38 pagesJIS G 3106 - NewMani Ma100% (2)
- UOP Adsorbent For Natural GasDocument3 pagesUOP Adsorbent For Natural Gas13201940Pas encore d'évaluation
- Solvent Free Green Synthesis of 5-Arylidine Barbituric Acid Derivatives Catalyzed by Copper Oxide NanoparticlesDocument6 pagesSolvent Free Green Synthesis of 5-Arylidine Barbituric Acid Derivatives Catalyzed by Copper Oxide NanoparticlesbaskhemPas encore d'évaluation
- Synthesis and Characterization of Some Novel Chalcone DerivativesDocument5 pagesSynthesis and Characterization of Some Novel Chalcone DerivativesRaoul WembePas encore d'évaluation
- Synthesis and Characterization of Cu(II), Ni(II), Mn(II) and Co(II) Complexes from Chalcone N(4)-methyl(phenyl)thiosemicarbazoneDocument6 pagesSynthesis and Characterization of Cu(II), Ni(II), Mn(II) and Co(II) Complexes from Chalcone N(4)-methyl(phenyl)thiosemicarbazoneKassimPas encore d'évaluation
- MacromolSymposia Template FERNANDADocument12 pagesMacromolSymposia Template FERNANDAFernanda DiasPas encore d'évaluation
- Synthesis of Oxazolidinone Phosphonate Derivatives, Part II: Jae-Min Hwang, Sung-Ho Yeom, and Kang-Yeoun JungDocument6 pagesSynthesis of Oxazolidinone Phosphonate Derivatives, Part II: Jae-Min Hwang, Sung-Ho Yeom, and Kang-Yeoun JungMuhammad Adzan AkbarPas encore d'évaluation
- Organic Chemistry: An Indian JournalDocument5 pagesOrganic Chemistry: An Indian Journalsnigdha shromaPas encore d'évaluation
- Org Lett 2006 8 2699 - CannabinoidsDocument4 pagesOrg Lett 2006 8 2699 - CannabinoidsFatty BhuwaneePas encore d'évaluation
- 6552-Article Text-11737-1-10-20210530Document11 pages6552-Article Text-11737-1-10-20210530Montazer MensoorPas encore d'évaluation
- Enantioselective Reduction of Tetralones Using Fungal CulturesDocument6 pagesEnantioselective Reduction of Tetralones Using Fungal CulturesGabriella GabyPas encore d'évaluation
- Artigo - Synthesis and Characterization of A New Mebendazole SaltDocument11 pagesArtigo - Synthesis and Characterization of A New Mebendazole SaltAline SantosPas encore d'évaluation
- Studies On Catalytic Spectrophotometry Using Polymer-Schiff Base Metal Complex As Mimetic EnzymeDocument9 pagesStudies On Catalytic Spectrophotometry Using Polymer-Schiff Base Metal Complex As Mimetic EnzymeAhmed AlmeraabiPas encore d'évaluation
- Synthesis of Using The Nanocomposite Catalyst: Biodiesel Mg/Al/Zn Hydrotalcite/SBA-15Document24 pagesSynthesis of Using The Nanocomposite Catalyst: Biodiesel Mg/Al/Zn Hydrotalcite/SBA-15Vikash ChandravanshiPas encore d'évaluation
- Naeimi 2015Document22 pagesNaeimi 2015Rushikesh G ParitPas encore d'évaluation
- Harada1996 PDFDocument7 pagesHarada1996 PDFSuper MoneyPas encore d'évaluation
- (Materials Science-Poland) Nanosized MoO3 As A Reusable Heterogeneous Catalyst For The Synthesis of 26-Bis (Benzylidene) CyclohexanonesDocument6 pages(Materials Science-Poland) Nanosized MoO3 As A Reusable Heterogeneous Catalyst For The Synthesis of 26-Bis (Benzylidene) CyclohexanonesMina ArshadPas encore d'évaluation
- Synthesis of high-molecular-weight carbazole polymers via Suzuki polycondensationDocument7 pagesSynthesis of high-molecular-weight carbazole polymers via Suzuki polycondensationSachin SahooPas encore d'évaluation
- Buchwald-Hartwig C-N Cross Coupling Reactions Catalyzed by A Pseudo-PincerDocument7 pagesBuchwald-Hartwig C-N Cross Coupling Reactions Catalyzed by A Pseudo-PincerAlberto ReyesPas encore d'évaluation
- Review of Literature V1Document8 pagesReview of Literature V1Naveena RathinavelPas encore d'évaluation
- Benzofuroxan (Benzo (1,2-c) 1,2,5-Oxadiazole N-Oxide) Derivatives As Potential Energetic Materials: Studies On Their Synthesis and PropertiesDocument22 pagesBenzofuroxan (Benzo (1,2-c) 1,2,5-Oxadiazole N-Oxide) Derivatives As Potential Energetic Materials: Studies On Their Synthesis and PropertiesJonas SarlauskasPas encore d'évaluation
- Yangbolun 200702 13Document4 pagesYangbolun 200702 13666667Pas encore d'évaluation
- Synthesis, Structural Characterization and Antimicrobial StudiesDocument4 pagesSynthesis, Structural Characterization and Antimicrobial StudiesManuel DíazPas encore d'évaluation
- Synthesis, Crystal Structure and NMR Assignments of 17 B-Acetoxy-4,5-Secoandrost-3-Yn-5-OneDocument5 pagesSynthesis, Crystal Structure and NMR Assignments of 17 B-Acetoxy-4,5-Secoandrost-3-Yn-5-Onetrikitraka3Pas encore d'évaluation
- Alexander A. Ezhov Et Al - Liquid-Crystalline Polymer Composites With CDS Nanorods: Structure and Optical PropertiesDocument13 pagesAlexander A. Ezhov Et Al - Liquid-Crystalline Polymer Composites With CDS Nanorods: Structure and Optical PropertiesYlpkasoPas encore d'évaluation
- ISSN 0974-4169: Research ArticleDocument5 pagesISSN 0974-4169: Research ArticleWalid EbaiedPas encore d'évaluation
- Oxido-And Dioxidovanadium V Complexes Wi PDFDocument10 pagesOxido-And Dioxidovanadium V Complexes Wi PDFAdnan Ahmed ChahalPas encore d'évaluation
- Jurnal Kimed 2Document8 pagesJurnal Kimed 2Wirna SelfiaPas encore d'évaluation
- Iraqi National Journal of Chemistry 2015; 15(2): Preparation of a new azo-chalcone ligandDocument23 pagesIraqi National Journal of Chemistry 2015; 15(2): Preparation of a new azo-chalcone ligandIon DerivolcovPas encore d'évaluation
- Benzimidazole MesogensDocument27 pagesBenzimidazole MesogensAbbas Washeel SalmanPas encore d'évaluation
- Docking, in Vitro DNA Binding, Cleavage, Anti-Proliferative and Antimicrobial Studies of O-Vanillin DerivativesDocument19 pagesDocking, in Vitro DNA Binding, Cleavage, Anti-Proliferative and Antimicrobial Studies of O-Vanillin Derivativespulimamidi SarithareddyPas encore d'évaluation
- Maniscript - TotallDocument17 pagesManiscript - Totall79ccpmh4cdPas encore d'évaluation
- Manson2011 Article PolyethyleneGlycolFunctionaliz PDFDocument7 pagesManson2011 Article PolyethyleneGlycolFunctionaliz PDFVaswati BiswasPas encore d'évaluation
- Crystal Structure and Behavior of Copper Complex in SolutionDocument8 pagesCrystal Structure and Behavior of Copper Complex in SolutionNathalia FlorenciaPas encore d'évaluation
- 295491Document7 pages295491khaliddarwish1962Pas encore d'évaluation
- Heterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaDocument5 pagesHeterogeneous Asymmetric Diels-Alder Reactions Using A Copper-Chiral Bis (Oxazoline) Complex Immobilized On Mesoporous SilicaJC Jane BarnesPas encore d'évaluation
- jo3c02343_si year 2024Document80 pagesjo3c02343_si year 2024pabitrapanigrahi02Pas encore d'évaluation
- Synthesis of Functional Amino Acids Bearing 1,3-Dithiane ModificationDocument4 pagesSynthesis of Functional Amino Acids Bearing 1,3-Dithiane ModificationHingryd RauenPas encore d'évaluation
- 10 1016@j Molstruc 2015 07 023Document6 pages10 1016@j Molstruc 2015 07 023Muhammad Abdur RokhimPas encore d'évaluation
- PropranololDocument6 pagesPropranololDaniel LawsonPas encore d'évaluation
- PNP Pourmousavi2017Document31 pagesPNP Pourmousavi2017sonPas encore d'évaluation
- Synthesis and Characterization of Schiff Base Ligands and Their Metal ComplexesDocument9 pagesSynthesis and Characterization of Schiff Base Ligands and Their Metal Complexesmaryam Saket OsgoueiPas encore d'évaluation
- Dieterle 1977Document9 pagesDieterle 1977Carolina Calvache LunaPas encore d'évaluation
- Cocrystal WorkDocument11 pagesCocrystal WorkMuhammad Al-HanafiPas encore d'évaluation
- Synthesis and Characterization of Some Vanadium (IV) and Vanadium (V) ComplexesDocument8 pagesSynthesis and Characterization of Some Vanadium (IV) and Vanadium (V) ComplexesCallsmetPas encore d'évaluation
- 1 s2.0 S0022286022014508 MainDocument12 pages1 s2.0 S0022286022014508 MainDavid V2'0Pas encore d'évaluation
- 3-NH Lig ThioureaDocument6 pages3-NH Lig ThioureaFarouk El NaggarPas encore d'évaluation
- CarmegliptinDocument12 pagesCarmegliptinQuân MinhPas encore d'évaluation
- Highly Efficient One-pot Synthesis, Antimicrobial and Docking Studies of Newer β-amino Carbonyl Derivatives Catalyzed by Silica Sulfuric AcidDocument14 pagesHighly Efficient One-pot Synthesis, Antimicrobial and Docking Studies of Newer β-amino Carbonyl Derivatives Catalyzed by Silica Sulfuric AcidnanoPas encore d'évaluation
- Vietnam Journal of Chemistry - A Facile Synthesis Characterization and Docking Studies of 2 Methyl 3Document9 pagesVietnam Journal of Chemistry - A Facile Synthesis Characterization and Docking Studies of 2 Methyl 3ramakrishna reddyPas encore d'évaluation
- Coordination Polymers Assembled From 3,3, 5,5 - Azobenzenetetracarboxylic Acid and Di Fferent Bis (Imidazole) Ligands With Varying FlexibilityDocument8 pagesCoordination Polymers Assembled From 3,3, 5,5 - Azobenzenetetracarboxylic Acid and Di Fferent Bis (Imidazole) Ligands With Varying Flexibilitykarthiche05Pas encore d'évaluation
- Synthesis & Characterization of Quinoxalines Using ENPFSA CatalystDocument29 pagesSynthesis & Characterization of Quinoxalines Using ENPFSA CatalystPrasada Rao Ch MMPas encore d'évaluation
- Highly Selective Oxalate - Membrane Electrode Based On (Cul) (Ac)Document10 pagesHighly Selective Oxalate - Membrane Electrode Based On (Cul) (Ac)Hani KhuludPas encore d'évaluation
- B120432 1279 PDFDocument6 pagesB120432 1279 PDFCarolina PalacioPas encore d'évaluation
- Synthesis of rhein derivative from aloe veraDocument5 pagesSynthesis of rhein derivative from aloe veraRajesh KumarPas encore d'évaluation
- JCPR 2012 4 2 1259 1265Document7 pagesJCPR 2012 4 2 1259 1265JuvansinhJadejaPas encore d'évaluation
- He Ravi 2007Document4 pagesHe Ravi 2007Vivek ShewalePas encore d'évaluation
- Silicotungstic Acid Supported Zirconia: An Effective Catalyst For Esterification ReactionDocument7 pagesSilicotungstic Acid Supported Zirconia: An Effective Catalyst For Esterification ReactionJenny CórdobaPas encore d'évaluation
- 1-s2.0-S0022286023020987-mainDocument9 pages1-s2.0-S0022286023020987-mainbellaoui.batounPas encore d'évaluation
- Sustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimeD'EverandSustainable synthesis of ciclopentene derivatives through multicomponent reactions in continuous flow regimePas encore d'évaluation
- Alogliptin - A Review of Its Use in Patients With Type 2 Diabetes MellitusDocument20 pagesAlogliptin - A Review of Its Use in Patients With Type 2 Diabetes Mellitus13201940Pas encore d'évaluation
- Types of Diabetes PillsDocument2 pagesTypes of Diabetes Pills13201940Pas encore d'évaluation
- Canagliflozin for Type 2 Diabetes TreatmentDocument7 pagesCanagliflozin for Type 2 Diabetes Treatment13201940Pas encore d'évaluation
- Gly. CarbonateDocument5 pagesGly. Carbonate13201940Pas encore d'évaluation
- Preparation of 3-MercaptopropionatesDocument12 pagesPreparation of 3-Mercaptopropionates13201940Pas encore d'évaluation
- Failed State US (Sociology)Document4 pagesFailed State US (Sociology)13201940Pas encore d'évaluation
- Indole AlkaloidsDocument286 pagesIndole Alkaloids13201940Pas encore d'évaluation
- Renewable Materials From FatsDocument30 pagesRenewable Materials From Fats13201940Pas encore d'évaluation
- Reduction of Aromatic Nitro Compounds With Activated IronDocument2 pagesReduction of Aromatic Nitro Compounds With Activated Iron13201940Pas encore d'évaluation
- CrystaSulf BrochureDocument4 pagesCrystaSulf Brochure13201940Pas encore d'évaluation
- Anthony Crasto Ugi ReactionDocument35 pagesAnthony Crasto Ugi ReactionAnthony Melvin Crasto Ph.DPas encore d'évaluation
- Binders For Coal BriquetsDocument56 pagesBinders For Coal Briquets13201940Pas encore d'évaluation
- Pulmonary Artery Hypertension (PAH) - BosentanDocument13 pagesPulmonary Artery Hypertension (PAH) - Bosentan13201940Pas encore d'évaluation
- Preparation of 3-MercaptopropionatesDocument12 pagesPreparation of 3-Mercaptopropionates13201940Pas encore d'évaluation
- Us 7326392Document12 pagesUs 732639213201940Pas encore d'évaluation
- Batch Processes in ChemistryDocument4 pagesBatch Processes in Chemistry13201940Pas encore d'évaluation
- An Overview MembraneDocument13 pagesAn Overview Membrane13201940Pas encore d'évaluation
- C1 Chemistry For The Production of Clean LiquidDocument101 pagesC1 Chemistry For The Production of Clean Liquid13201940Pas encore d'évaluation
- Technical Challenges UOPDocument23 pagesTechnical Challenges UOP13201940100% (2)
- Advances in The Synthesis of GlyceridesDocument24 pagesAdvances in The Synthesis of Glycerides13201940Pas encore d'évaluation
- Us4745207 HCNDocument5 pagesUs4745207 HCN13201940Pas encore d'évaluation
- ABB SR 60 Years of HVDC - 72dpi PDFDocument72 pagesABB SR 60 Years of HVDC - 72dpi PDFroyclhorPas encore d'évaluation
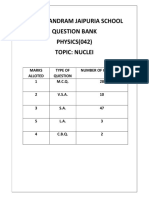
- Nuclei QB XiiDocument23 pagesNuclei QB XiiToshani GuptaPas encore d'évaluation
- Flywheels May SulatDocument3 pagesFlywheels May SulatRonniePas encore d'évaluation
- Subd - of Lot 3-c.ldcDocument4 pagesSubd - of Lot 3-c.ldcYza RoblesPas encore d'évaluation
- Week 1 ScienceDocument38 pagesWeek 1 ScienceEyphrille UmandapPas encore d'évaluation
- Charging Processes ExplainedDocument4 pagesCharging Processes ExplainedMa'am Joana Joy PalomaresPas encore d'évaluation
- H 103 - ISO - Rev10 - INGDocument1 pageH 103 - ISO - Rev10 - INGandersmorais86Pas encore d'évaluation
- Thermodynamics of Heating PDFDocument19 pagesThermodynamics of Heating PDFMatias MuñozPas encore d'évaluation
- Unit & DimensionsDocument9 pagesUnit & DimensionsRandhir SinghPas encore d'évaluation
- Answer All Questions: Igcse/O Level Physics Light ExerciseDocument34 pagesAnswer All Questions: Igcse/O Level Physics Light ExerciseNasser AlsowyanPas encore d'évaluation
- Bidwell 2005Document6 pagesBidwell 2005Arturo GBPas encore d'évaluation
- Intensive and Extensive Properties Crowther-Robitaille 2019Document6 pagesIntensive and Extensive Properties Crowther-Robitaille 2019provocator74Pas encore d'évaluation
- Mechanical EngineeringDocument19 pagesMechanical EngineeringhenoksolPas encore d'évaluation
- Gribs PacketDocument10 pagesGribs Packetapi-213645632Pas encore d'évaluation
- Magnetism NotesDocument4 pagesMagnetism NotesvenuPas encore d'évaluation
- B.Tech. 1st Term Q15 PDFDocument10 pagesB.Tech. 1st Term Q15 PDFDEBAPRASAD PALPas encore d'évaluation
- SM04 Poster 82Document6 pagesSM04 Poster 82lkamalPas encore d'évaluation
- Reserves Estimation For A Coal Bed Methane Well PETSOC-03-11-01-PDocument6 pagesReserves Estimation For A Coal Bed Methane Well PETSOC-03-11-01-Psaladinayubi1234Pas encore d'évaluation
- POLE FOUNDATION ANALYSISDocument2 pagesPOLE FOUNDATION ANALYSISPrabuVijayPas encore d'évaluation
- Brochure CTS-9005Document4 pagesBrochure CTS-9005Dika AnggaraPas encore d'évaluation
- Year 10 Trigonometry 2Document4 pagesYear 10 Trigonometry 2Jack LagerPas encore d'évaluation
- Lesson 3-F5 PhysicsDocument14 pagesLesson 3-F5 PhysicsCheng WLPas encore d'évaluation
- Thomeer Swanson Type Curve MatchingDocument60 pagesThomeer Swanson Type Curve MatchingDeepblue09Pas encore d'évaluation