Académique Documents
Professionnel Documents
Culture Documents
Cap 1
Transféré par
Hernán Reales VegaTitre original
Copyright
Formats disponibles
Partager ce document
Partager ou intégrer le document
Avez-vous trouvé ce document utile ?
Ce contenu est-il inapproprié ?
Signaler ce documentDroits d'auteur :
Formats disponibles
Cap 1
Transféré par
Hernán Reales VegaDroits d'auteur :
Formats disponibles
CAPITULO 1 Adaptaciones celulares, lesin celular y muerte celular INTRODUCCIN A LA PATOLOGA ASPECTOS GENERALES DE LA RESPUESTA CELULAR AL ESTRS Y A L OS ESTMULOS
NOCIVOS ADAPTACIONES CELULARES DEL CRECIMIENTO Y DIFERENCIACIN Hiperpl asia Hiperplasia fisiolgica Hiperplasia patolgica Hipertrofia Atrofia Metaplasia A SPECTOS GENERALES DE LA LESIN Y MUERTE CELULARES CAUSAS DE LESIN CELULAR MECANISMO S DE LESIN CELULAR Deplecin de ATP Dao mitocondrial Aflujo del calcio intracelular y prdida de la homeostasia del calcio Acumulacin de radicales libres derivados de oxgeno (estrs oxidativo) Defectos en la permeabilidad de membrana LESIN CELULAR REV ERSIBLE E IRREVERSIBLE MORFOLOGA DE LA LESIN Y NECROSIS CELULARES Lesin reversible Necrosis EJEMPLOS DE LESIN Y NECROSIS CELULARES Lesin isqumica e hipxica Lesin de isquemia-rep erfusin Lesin qumica APOPTOSIS Causas de apoptosis Apoptosis en situaciones fisiolgi cas Apoptosis en situaciones patolgicas Caractersticas bioqumicas de la apoptosis M ecanismos de la apoptosis Ejemplos de apoptosis RESPUESTAS SUBCELULARES A LA LES IN Catabolismo lisosomal Induccin (hipertrofia) del retculo endoplsmico liso Alterac iones mitocondriales Anomalas citoesquelticas ACMULOS INTRACELULARES Lpidos Esteatos is (cambio graso) Colesterol y esteres de colesterol Protenas Cambio hialino Glucg eno Pigmentos CALCIFICACIN PATOLGICA Calcificacin distrfica Calcificacin metastsica EN VEJECIMIENTO CELULAR
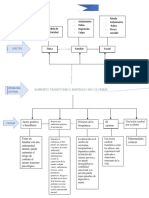
cortantes como la lesin celular para definii ractersticas clnicas de la enfermedad. setos generales de la respu lar al estrs y a los estimule vos la normal est limitada a un abanico bastar
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 5 CLULA NORMAL (homeostasia) Estrs, demanda aumentada, Estmulo agresor ADAPTACIN Incapacidad de adaptacin AGRESIN ->. CELULAR V : . . . : FIGURA 1-1 Estadios en la respuesta celular al estrs y los estmulos nocivos.
es una respuesta adaptativa en la que existe una disminucin en el tamao y funcin de las clulas. Si se sobrepasan los lmites de la respuesta adaptativa a un estmulo, o en ciertas situaciones cuando la clula est expuesta a un agente lesivo o a estrs, se sucede una secuencia de acontecimientos que se denomina inconcretamente como lesin celular. La lesin celular es reversible hasta cierto punto, pero si el estmul o persiste o es lo bastante intenso desde el principio la clula alcanza un punto d e no retorno y sufre lesin celular irreversible y, Adaptaciones celulares finalmen te, muerte celular. La adaptacin, lesin reversible^ muerte celular pueden consider arse estadios de deterioro progresivo de del crecimiento y diferenciacin la funcin y estructura normales de la clula (ver Fig. 1-1). Por Las clulas responden a un a umento de la demanda y del estmuejemplo, en respuesta al aumento de cargas hemodi nmicas, el lo externo con hiperplasia o hipertrofia, y responden con atrofia a msc ulo cardaco primero aumenta de tamao, una forma de un suministro reducido de nutri entes y factores de crecimiento. adaptacin. Si el riego sanguneo al miocardio es i nsuficiente para En algunas situaciones, las clulas cambian de un tipo a otro, un hacer frente a la demanda, el msculo se lesiona reversiblemente proceso denomina do metaplasia. Existen numerosos mecanismos y,finalmente,acaece la muerte celula r (Fig. 1-2). celulares para las adaptaciones celulares. Algunas adaptaciones La muerte celular, el resultado ltimo de la lesin celular, es uno estn inducidas por el estmulo directo de las clulas por factores producidos por las mismas clulas resp ondedoras o por otras de los acontecimientos ms cruciales en la evolucin de la enf erclulas circundantes. Otras se deben a la activacin de diversos medad de cualquie r tejido u rgano. Es el resultado de diversas receptores celulares de superficie y de las vas de sealizacin subcausas, incluyendo isquemia (falta de riego sanguneo), infeccin, siguientes. Las adaptaciones pueden asociarse con la induccin de toxina s y reacciones inmunitarias. Adems, la muerte celular es una nueva sntesis proteic a por las clulas diana, como ocurre en una parte normal y esencial de la embriogne sis, el desarrollo de la respuesta de las clulas musculares al aumento de la dema nda rganos y el mantenimiento de la homeostasia, y es el objetivo fsica, y la indu ccin de proliferacin celular, como sucede en las de la teraputica del cncer. Hay dos patrones principales de muerte celular: la necrosis y la apoptosis. La necrosis es el tipo respuestas del endometrio a estrgenos. Las adaptaciones pueden implic ar tambin un cambio celular desde la produccin de un de muerte celular que ocurre despus de tipos de estrs anormatipo de protenas a otro o la hiperproduccin marcada d e una les tales como isquemia y lesin qumica, y siempre es patolgica. La apoptosis ocurre cuando una clula muere por la activacin de protena; tal es el caso de las clu las que producen diversos tipos de colgeno y protenas de matriz extracelular en la inflamacin un programa suicida controlado internamente. Est diseada crnica yfibrosi s(Captulos 2 y 3). para eliminar clulas indeseadas durante la embriognesis y en TABLA 1-1 Naturaleza e intensidad del estmulo lesivo Estmulo fisiolgico alterado: A umento de la demanda, aumento del estmulo trfico (p. ej., factores de crecimiento, hormonas) Disminucin de nutrientes, estimulacin Irritacin crnica (qumica o fsica) rte reducido de oxgeno; lesin qumica; infeccin microbiana: Aguda y autolimitada Prog resiva y grave (incluyendo dao del DNA) Lesin crnica leve Respuestas celulares a la lesin Respuesta celular Adaptaciones celulares: Hiperplasia, hipertrofia Atrofia Metaplasia Dao celular: Lesin aguda reversible Lesin irreversible - muerte celular Necrosis Apoptosis Alteraciones subcelulares en varias organelas Acumulaciones i
ntracelulares; calcificaciones Envejecimiento celular varios procesosfisiolgicos,tales como la involucin de tejidos respondedores a horm onas cuando se retira la hormona. Tambin ocurre en ciertas afecciones patolgicas, cuando las clulas se daan ms all de su reparacin, y especialmente si el dao afecta al DNA nuclear de la clula. Ms adelante en este captulo volveremos a una exposicin deta llada de esas vas de muerte celular. Diferentes tipos de estrs pueden inducir camb ios celulares y tisulares distintos de la adaptacin, lesin celular y muerte (ver T abla 1-1). Las clulas expuestas a estmulos subletales o crnicos pueden no daarse, si no mostrar diversas alteraciones subcelulares. Los trastornos metablicos en las cl ulas pueden asociarse con acmulos intracelulares de diversas sustancias, incluyen do protenas, lpidos e hidratos de carbono. El calcio se deposita a menudo en los s itios de muerte celular, dando lugar a calcificacin patolgica. Finalmente, el enve jecimiento celular tambin se acompaa de cambios morfolgicos y funcionales caracterst icos. En este captulo primeramente exponemos cmo se adaptan las clulas al estrs, y d espus las causas, mecanismos y consecuencias de diversas formas de dao celular agu do, incluyendo lesin celular y muerte celular. Concluimos con las alteraciones su bcelulares inducidas por los estmulos subletales, acumulaciones intracelulares, c alcificacin patolgica y envejecimiento celular. Alteraciones metablicas, genticas o adquiridas Duracin prolongada de la vida con le sin subletal acumulativa
6 UNIDAD I Patologa general Miocito normal Miocito reversiblemente lesionado FIGURA 1-2 Relaciones entre las clulas miocrdicas normales, adaptadas, reversiblem ente lesionadas y muertas. La adaptacin celular mostrada aqu es la hipertrofia, y el tipo de muerte celular la necrosis isqumica. En el miocardio reversiblemente l esionado, generalmente hay efectos slo funcionales, sin ningn cambio abiertamente aparente macroscpico ni siquiera microscpico. En el ejemplo de la hipertrofia miocr dica, el ventrculo izquierdo tiene un grosor de ms de 2 cm (lo normal es de 1 a 1, 5 cm). En la muestra con necrosis, el rea clara transmural en el ventrculo izquier do posterolateral representa un infarto de miocardio agudo. Las tres secciones t ransversas se han teido con cloruro de trifeniltetrazolio, un sustrato enzimtico q ue colorea de magenta el miocardio viable. La ausencia de tincin se debe a derram e enzimtico tras la muerte celular. a, y su hgado era devorado todos los das por un buitre para regenerarse de nuevo ca da noche1. El modelo experimental de La hiperplasia es un aumento en el nmero de clulas en un rgahepatectoma parcial ha sido especialmente til para examinar los no o tejido, dando lugar habitualmente a un aumento del volumecanismos que estimulan la proliferacin de las clulas heptimen del rgano o tejido. Aunque la hiperplasia y la hipertrofia cas residuales y la regeneracin del hgado (Captulo 3). Probason dos procesos distintos, frecuentemente ambos ocurren junblemente, mecanismos similar es estn implicados en otras situatos, y pueden desencadenarse por el mismo estmulo externo. Por ciones cuando el tejido restante crece para compensar la prdida eje mplo, el crecimiento inducido hormonalmente en el tero parcial de tejido (p. ej., tras nefrectoma unilateral, cuando en el implica a la vez un nmero aumentado de cl ulas musculares y rion restante se produce una hiperplasia compensadora). clulas e piteliales y el agrandamiento de esas clulas. La hiperplaMecanismos de hiperplasi a. La hiperplasia generalmente se sia tiene lugar si la poblacin celular es capaz de sintetizar DNA, debe a una produccin local aumentada de factores de crecimien permitiendo as la divisin mittica; en contraste, la hipertrofia to, niveles aumenta dos de receptores de factor de crecimiento en implica el agrandamiento celular s in divisin de la clula. La las clulas respondedoras, o activacin de una determinada va de hiperplasia puede serfisiolgicao patolgica. sealizacin intracelular. Todos esto s cambios dan lugar a la produccin de factores de transcripcin que activan muchos genes celulares, incluyendo genes que codifican factores de crecimiento, Hiperpl asia fisiolgica receptores de factores de crecimiento y reguladores del ciclo cel u2 La hiperplasiafisiolgicapuede dividirse en: (1) hiperplasia hor- lar, con el r esultado neto de una proliferacin celular . En la hiperplasia hormonal, las propi as hormonas actan como factomonal, que aumenta la capacidad funcional de un tejid o cuando se necesita, y (2) hiperplasia compensadora, que aumenta la masa res de crecimiento y desencadenan la transcripcin de diversos genes celulares. La fuent e de factores de crecimiento en la hipertisular tras el dao o reseccin parcial. El mejor ejemplo de hiperplasia compensadora y el estmulo para la produccin de estos plasia hormonal es la proliferacin del epitelio glandular de la factores estn peo r definidos. El aumento en la masa tisular tras mama femenina en la pubertad y d urante el embarazo y la hiperalgunos tipos de prdida celular se consigue no slo po r prolifeplasia fisiolgica que ocurre en el tero grvido. La ilustracin racin de las r estantes clulas sino tambin por el desarrollo de clsica de hiperplasia compensadora deriva del mito de Prometeo, nuevas clulas a partir de clulas madreiA. Por ejempl o, en el hgaque muestra que los griegos antiguos reconocan la capacidad de do, las clulas madre intrahepticas no desempean un papel regeneracin del hgado. Como castigo por haber robado el secreimportante en la hiperplasia que tiene lugar tras hepa tectoma to del fuego a los dioses, Prometeo fue encadenado a una monta-
HIPERPLASIA
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular pero pueden par ticipar en la regeneracin tras ciertas formas de lesin heptica, tales como hepatiti s crnica, en las cuales est comprometida la capacidad proliferativa de los hepatoc itos. Datos recientes de observaciones clnicas y estudios experimentales han demo strado que la mdula sea contiene clulas madre que pueden ser capaces de dar lugar a diversos tipos de clulas diferenciadas, especializadas, incluyendo algunas clulas hepticas5. Estas observaciones ilustran la plasticidad de las clulas madre del ad ulto y sugieren la capacidad potencial de repoblar los tejidos daados con clulas m adre derivadas de la mdula sea. Volveremos a abordar el tema de las clulas madre, s u biologa y su importancia clnica en el Captulo 3. Hiperplasia patolgica 7 La mayora de las formas de hiperplasia patolgica estn producidas por un estmulo horm onal excesivo o factores de crecimiento que actan sobre clulas diana. La hiperplas ia endometrial es un ejemplo de hiperplasia anormal inducida por hormonas. Tras un perodo menstrual normal, hay un estallido rpido de actividad proliferativa que se estimula por hormonas hipofisarias y estrgeno ovrico. Se detiene con los nivele s crecientes de progesterona, aproximadamente a los 10 a 14 das antes del esperad o perodo menstrual. Sin embargo, en algunos casos, el equilibrio entre estrgeno y progesterona est alterado. Esto da lugar a aumentos absolutos o relativos de estrg eno, con la hiperplasia subsiguiente de las glndulas endometriales. Esta forma de hiperplasia es El crecimientofisiolgicomasivo del tero durante el embarauna causa frecuente de hemorragia menstrual anormal. La hiperzo es un buen ejemplo del au mento del tamao de un rgano plasia prosttica benigna es otro ejemplo frecuente de h iperplasia inducido por hormonas, que es el resultado tanto de hipertrofia patolg ica inducida por respuestas a hormonas, en este caso como hiperplasia (Fig. 1-3A ). La hipertrofia celular se estimula andrgenos. Aunque estas formas de hiperplas ia son anormales, por hormonas estrognicas que actan sobre los receptores estroel proceso permanece controlado, ya que la hiperplasia regresa gnicos del msculo liso , dando lugar finalmente a una sntesis si se elimina el estmulo hormonal. Como se expone en el Captuaumentada de las protenas del msculo liso y a un aumento en el lo 7, es esta respuesta a los mecanismos de control regulador nortamao celular (Fig . 1-3B). De igual forma, la prolactina y males lo que distingue a las hiperplasi as benignas patolgicas del el estrgeno producen hipertrofia de las mamas durante l a laccncer, en el cual los mecanismos de control de crecimiento se tancia. stos so n ejemplos de hipertrofiafisiolgicainducida por han hecho defectuosos. Sin embarg o, la hiperplasia patolgica estmulo hormonal. constituye un terreno frtil en el cua l, puede surgir posteriormente la Aunque el punto de vista tradicional sobre el msculo cardaproliferacin cancerosa. As pues, las pacientes con hiperplasia de co y e squeltico es que estos tejidos son incapaces de proliferacin endometrio tienen un riesgo aumentado de desarrollar cncer y, por lo tanto, su agrandamiento es el res ultado enteramente de de endometrio (Captulo 22). la hipertrofia, los datos recie ntes sugieren que incluso estos tipos La hiperplasia tambin es una respuesta impo rtante de las clucelulares son capaces de una proliferacin limitada as como de las del tejido conectivo en la curacin de heridas, en las que la una repoblacin a part ir de precursores6. Este punto de vista proliferacin defibroblastosy vasos sangune os ayuda a la repasubraya el concepto, mencionado anteriormente, de que la hiper racin (Captulo 3). En estas circunstancias, los factores de creciplasia y la hiper trofia a menudo ocurren concomitantemente miento son responsables de la hiperpla sia. El estmulo mediante durante las respuestas de los tejidos y rganos ante un au mento factores de crecimiento tambin est implicado en la hiperplasia de estrs y prdi da celular. que se asocia con ciertas infecciones vricas, tales como papilomaMeca nismos de hipertrofia. La mayora de nuestro conocivirus, que producen verrugas en la piel y una diversidad de lesiomiento sobre la hipertrofia se basa en estudio s del corazn. Los nes de las mucosas compuestas de masas de epitelio hiperplsico. mecanismos de la hipertrofia del msculo cardaco implican muchas vas de transduccin d e seal, dando lugar a la induccin de un nmero de genes que, a su vez, estimulan la sntesis de HIPERTROFIA numerosas protenas celulares (Fig. 1-4)7'8. Los genes que s e induLa hipertrofia se refiere a un aumento en el tamao de las clulas, lo durante
la hipertrofia incluyen los que codifican factores de cen que da lugar a un aum ento en el tamao del rgano. As pues, el transcripcin (tales como c-fos, c-jun); fact ores de crecimiento rgano hipertrofiado no tiene nuevas clulas, sino solamente (TG B-f$, factores de crecimiento de tipo insulina-1 [IGF-1], facclulas mayores. El t amao aumentado de las clulas no se debe a tor de crecimientofibroblstico),y agentes vasoactivos (agonistas hinchazn celular sino a la sntesis de ms componentes estruc tua-adrenrgicos, endotelina-1 y angiotensina II). Estos factores se rales. Como s e dijo antes, las clulas capaces de divisin pueden abordan en detalle en el Captulo 3. Tambin puede haber un responder al estrs con hiperplasia y tambin con hipertrof ia, cambio de las protenas contrctiles del adulto a formas fetales o mientras que en las clulas que no se dividen (p. ej., fibras miocr- neonatales. Por ejemplo, du rante la hipertrofia muscular, la cadedicas) ocurre hipertrofia. Los ncleos en la s clulas hipertrofiadas na pesada de a-miosina es sustituida por la forma |3 de l a cadena pueden tener un mayor contenido en DNA que las clulas norpesada de miosi na, lo que da lugar a una disminucin de la actimales, probablemente porque las clulas se detienen en el ciclo celular sin llevar a cabo la mitosis. La hipertrofia puede ser fisiolgica o patolgica y est producida por un aumento de la demanda funcional o por estimulacin hormonal especfica. Las clulas del msculo estriado en el corazn y en los msculos esquelticos son capaces de u na hipertrofia tremenda, quiz porque no pueden adaptarse adecuadamente al aumento de las demandas metablicas por la divisin mittica y la produccin de ms clulas para co mpartir el trabajo. El estmulo ms frecuente para la hipertrofia del msculo es el au mento de su trabajo. Por ejemplo, los msculos voluminosos de los culturistas impl icados en el bombeo de hierro son el resultado de un aumento en el tamao de las fib ras musculares individuales en respuesta al aumento de la demanda. As, la carga d e trabajo est compartida por una mayor masa de componentes celulares, y a cada fi bra muscular se le ahorra un exceso de trabajo y de esta manera escapa a la lesin . La clula muscular agrandada consigue un nuevo equilibrio, permitindole funcionar a un mayor nivel de actividad. En el corazn, el estmulo para la hipertrofia habit ualmente es la sobrecarga hemodinmica crnica, como consecuencia de la hipertensin o de vlvulas defectuosas. Ocurre una sntesis de ms protenas y filamentos, consiguindos e un equilibrio entre la demanda y la capacidad funcional de la clula. El mayor nm ero de miofilamentos por clula permite un aumento de carga de trabajo con un nive l de actividad metablica por unidad de volumen celular no diferente del que sopor ta una clula normal.
8 UNIDAD I '$ Patologa general FIGURA 1-3 Hipertrofia fisiolgica del tero durante el embarazo. A, apariencia macr oscpica de un tero normal (derecha) y un tero grvido (extrado por hemorragia posparto ) {izquierda). B, pequeas clulas fusiformes del msculo liso uterino normal (izquier da) en comparacin con clulas grandes redondeadas en un tero grvido (derecha). AGONISTAS: RECEPTORES Hormonas a-adrenrgicas Angiotensina I Endotelina Factores de crecimiento Distensin mecnica FIGURA 1-4 Cambios en la expresin de genes seleccionados y protenas durante la hipertrofia de miocardio. vidad de la adenosintrifosfatasa (ATPasa) de la miosina y a una contraccin ms lent a, energticamente ms econmica. Adems, algunos genes que se expresan solamente durant e el desarrollo precoz se vuelven a expresar en las clulas hipertrficas, y los pro ductos de esos genes participan en la respuesta celular al estrs. Por ejemplo, en el corazn embrionario, el gen para el factor natriurtico auricular (FNA) se expre sa en la aurcula y en el ventrculo. Tras el nacimiento, la expresin ventricular del gen est disminuida. Sin embargo, la hipertrofia cardaca se asocia con la reinducc in de la expresin del FNA9. El FNA es una hormona peptdica que produce secrecin de s al por el rion, disminuye el volumen y la presin sangunea y, por lo tanto, sirve pa ra reducir la carga hemodinmica. Cules son los desencadenantes de la hipertrofia y de estos cambios en la expresin d el gen? En el corazn existen al menos dos grupos de seales: los desencadenantes me cnicos, tales como la distensin, y los desencadenantes trficos, tales como los fact ores de crecimiento polipeptdicos (IGF-1) y los agentes vasoactivos (angiotensina II, agonistas ct-adrenrgicos). Los modelos actuales sugieren que los factores de crecimiento o los agentes vasoactivos producidos por clulas cardacas no musculare s o por los mismos miocitos en respuesta al estrs hemodinmico, estimulan la expres in de diversos genes, dando lugar a hipertrofia mioctica. El tamao de las clulas est regulado por nutrientes y seales ambientales e implica varias vas de transduccin de seales que se estn desentraando 10 .
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular Cualquiera que sea el mecanismo exacto de la hipertrofia cardiaca, finalmente alcanza un lmite ms all del cual el agrandamiento de la masa muscular ya no es capaz de compensar el aumento de la carga y se sigue de insuficiencia cardaca. En este estadio, ocurre n varios cambios degenerativos en las fibras miocrdicas, de los cuales los ms impo rtantes son la lisis y la prdida de los elementos contrctiles miofibrilares. La mu erte del miocito puede ocurrir por apoptosis o por necrosis". Los factores limit adores de la hipertrofia continuada y las causas de disfuncin cardaca se conocen p oco; pueden deberse a una limitacin del suministro vascular a lasfibrasaumentadas de tamao, a capacidades oxidativas disminuidas de la mitocondria, a alteraciones en la sntesis proteica y la degradacin, o a alteraciones citoesquelticas. 9
Prdida de inervacin {atrofiapor denervacin). La funcin normal del msculo esqueltico d pende de su suministro neurolgico. El dao en los nervios da lugar a una atrofia rpi da de las fibras musculares dirigidas por esos nervios (Captulo 27). Riego sangune o disminuido. Una disminucin en el riego sanguneo (isquemia) en un tejido resultad o de una enfermedad arterial oclusiva da lugar a atrofia del tejido debida a una prdida celular progresiva. En la vida adulta tarda, el cerebro sufre una atrofia progresiva, presumiblemente porque la aterosclerosis restringe su riego sanguneo (Fig. 1-5). Nutricin inadecuada. Una gran malnutricin proteicocalrica (marasmo) se asocia con el uso del msculo esqueltico como fuente de energa despus de haberse agot ado otras reservas como los almacenes adiposos. Esto da lugar a un gran consumo muscular {caquexia). La caquexia tambin se ve en ATROFIA pacientes con enfermedad es inflamatorias crnicas y cncer. Se La disminucin en el tamao de la clula por prdida de sustancia piensa que en las primeras, la hiperproduccin crnica de la celular se conoce como atrofia. Representa una forma de respuesta citocina inflamatoria fa ctor de necrosis tumoral (TNF) es la responsable de la supresin del apetito y de la atrofia muscular. adaptativa y puede culminar en la muerte celular. Cuando es t implicado un nmero suficiente de clulas, todo el tejido o el rga Prdida del estmulo ndocrino. Muchas glndulas endocrino disminuye de tamao, o se hace atrfico. La atrof ia puede ser nas, la mama y los rganos reproductores dependen del estfisiolgica o p atolgica. La atrofiafisiolgicaes comn durante el mulo endocrino para el metabolismo y funcin normales. La principio del desarrollo. Algunas estructuras embrionarias , tales prdida de estmulo estrognico tras la menopausia da lugar a como la notocord a y el conducto tireogloso, sufren atrofia duranla atrofia fisiolgica del endomet rio, el epitelio vaginal y la te el desarrollo fetal. El tero disminuye de tamao p oco despus mama. del parto, y sta es una forma de atrofiafisiolgica.La atrofia pato Envejecimiento {atrofia senil). El proceso de envejecimiento lgica depende de la causa subyacente y puede ser localizada se asocia con prdida celular, que se ve tp icamente en los tejio generalizada. Las causas comunes de atrofia son las siguie ntes: dos que contienen clulas permanentes, particularmente el corazn y el cerebro . Carga de trabajo disminuida {atrofia por desuso). Cuando un Presin. La compresin tisular durante algn perodo de tiempo puede producir atrofia. Un tumor benigno en crecimiembro roto se inmoviliza con una escayola o cuando el miento puede produ cir atrofia en los tejidos circundantes compaciente est sujeto a un reposo comple to en cama, rpidaprimidos. La atrofia en esta situacin es, probablemente, el mente se sigue de una atrofia muscular esqueltica. La dismiresultado de cambios isqumic os producidos por el comprominucin rpida inicial en el tamao celular es reversible una vez so del riego sanguneo a esos tejidos por la masa en expansin. que se retom a la actividad. Con la falta de uso ms prolongada las fibras musculares esqueltica s disminuyen en nmero as Los cambios celulares fundamentales asociados con atrofia son como en tamao; esta atrofia puede acompaarse de un idnticos en todas estas cir cunstancias, representando una retiraaumento en la resorcin sea, dando lugar a ost eoporosis por da celular a un tamao menor en el cual la supervivencia todava falta de uso. FIGURA 1-5 A, atrofia del cerebro en un hombre de 82 aos con enfermedad ateroscle rtica. La atrofia cerebral se debe al envejecimiento y a la disminucin del riego s
anguneo. Se han extirpado las meninges. B, cerebro normal de un hombre de 36 aos. Ntese que la prdida de sustancia cerebral estrecha las circunvoluciones y ensancha los surcos.
10 UNIDAD I # Patologa general es posible. La atrofia es el resultado de una reduccin en los componentes estruct urales de la clula. En el msculo atrfico, las clulas contienen menos mitocondrias y miofilamentos y una cantidad reducida de retculo endoplsmico. Ajusfando el volumen celular a los menores niveles de riego sanguneo, nutricin o estmulo trfico se consi gue un nuevo equilibrio. Aunque las clulas atrficas pueden tener una funcin disminu ida, no estn muerMembrana tas. Sin embargo, la atrofia puede progresar hasta el p unto en el basal que las clulas estn lesionadas y mueren. En los tejidos isqumicos, si el riego sanguneo es inadecuado incluso para mantener la vida de las clulas re tradas, pueden sobrevenir la lesin y la muerte celular. Adems, se puede inducir la apoptosis por las mismas seales que producen la atrofia y, de esta manera, puede contribuir a la prdida de masa orgnica. Por ejemplo, la apoptosis contribuye a la regresin de rganos endocrinos tras la retirada hormonal. Epitelio Clulas columnar de reserva normal Metaplasia escamosa Mecanismos de atrofia. Los mecanismos bioqumicos responsables de atrofia no se co nocen completamente pero quizs afectan al equilibrio entre la sntesis proteica y s u degradacin. Un aumento de la degradacin proteica desempea, probablemente, un pape l clave en la atrofia. Las clulas de mamferos contienen mltiples sistemas proteoltic os que tienen funciones diferentes. Los lisosomas contienen hidrolasas acidas (p . ej., catepsinas) y otras enzimas que degradan las protenas captadas por endocit osis del entorno extracelular y de la superficie celular, as como algunos compone ntes celulares. La va ubicuitina-proteosoma es responsable de la degradacin de muc has protenas citoslicas y FIGURA 1-6 Metaplasia. A, diagrama esquemtico de metaplas ia nucleares12. Las protenas que se degradan por este proceso se columnar a escam osa. B, transformacin metaplsica del epitelio conjugan primeramente con la ubicuit ina y despus se degradan escamoso estratificado del esfago {izquierda) a epitelio columnar maduro (la denominada metaplasia de Barrett). dentro de una gran organe la citoplasmtica proteoltica denominada proteosoma. Se piensa que esta va es la res ponsable de la protelisis acelerada que se ve en una variedad de afecciones catabl icas, incluyendo la caquexia cancergena. Las hormonas, partiepiteliales columnare s ciliadas normales de la trquea y de los cularmente los glucocorticoides y la ho rmona tiroidea, estimulan bronquios estn, a menudo, sustituidas localmente o difu samenla degradacin proteica mediada por proteosoma; la insulina se te por clulas e piteliales escamosas estratificadas. Los clculos en opone a esas acciones. Adicio nalmente, las citocinas, como el faclos conductos excretores de las glndulas sali vales, pncreas o tor de necrosis tumoral (TNF), son capaces de aumentar la procon ductos biliares pueden producir sustitucin del epitelio telisis muscular mediante este mecanismo. columnar secretor normal por epitelio escamoso estratificado no funcional. Una deficiencia en vitamina A (cido retinoico) induEn muchas situacion es, la atrofia se acompaa tambin de un aumento marcado en el nmero de vacuolas auto fgicas. stas son ce metaplasia escamosa en el epitelio respiratorio, y el exceso e n vacuolas ligadas a la membrana dentro de la clula que contienen vitamina A supr ime la queratinizacin (Captulo 9). En todos estos casos, el ms resistente epitelio escamoso estratificado es fragmentos de componentes celulares (p. ej., mitocondr ias, recapaz de sobrevivir en circunstancias en que el ms frgil epitelio tculo endo plsmico) que estn destinados a su destruccin en los cuales los lisosomas descargan sus contenidos hidrolticos. A con- columnar especializado probablemente habra sucu mbido. Aunque las clulas escamosas metaplsicas del tracto respiratorio, por tinuac in, los componentes celulares se digieren. Algunos de estos ejemplo, sean capaces de sobrevivir, se pierde un mecanismo proresiduos dentro de la vacuola autofgica pueden resistir la digestector importante -la secrecin mucosa-. Por tanto, la me taplasia tin y persistir como cuerpos residuales ligados a la membrana epitelial es una espada de doble filo y, en la mayora de casos, que pueden permanecer como
sarcfagos en el citoplasma. Un ejemplo de estos cuerpos residuales son los granul os de lipofuscina,representa un cambio indeseable. Ms an si persisten las influenc ias que predisponen a la metaplasia pueden inducir transformacin que se abordan d espus en este captulo. Cuando estn presentes neoplsica en el epitelio metaplsico. As l a presentacin comn de en cantidades suficientes, imparten un cambio de coloracin cnc er en el tracto respiratorio est formada por clulas escamomarrn al tejido (atrofiap arda). sas, que surgen en reas de metaplasia del epitelio columnar normal en epit elio escamoso. METAPLASIA La metaplasia del tipo escamoso a columnar tambin puede ocuLa metaplasia es un cambio reversible por el cual una clula de rrir, como en el esfago de Barrett, en el cual el epitelio escamoso tipo adulto (epitelial o me senquimal) se sustituye por otro tipo esofgico se sustituye por clulas columnares semejantes a las celular adulto13. Puede representar una sustitucin adaptativa in testinales bajo la influencia de jugo gstrico refluido (Fig. 1-6B). de clulas que son sensibles al estrs por tipos celulares mejor En esas reas pueden surgir cnceres , y tpicamente son capacitados para soportar el ambiente adverso. (adeno)carcinom as glandulares (Captulo 17). La metaplasia epitelial ms frecuente es de columnar a escamoLa metaplasia del tejido conectivo es la formacin de cartlago, sa (Fig. 1-6 A), como ocurre en el tracto respiratorio como reshueso o tejido adiposo (tejido s mesenquimales) en tejidos que norpuesta a la irritacin crnica. En el fumador hab itual, las clulas malmente no contienen esos elementos. Por ejemplo, la formacin
CAPTULO 1 Adaptacionas cglulares, lesin celular y muerte celular de hueso en el msc ulo, denominada miositis osificante, ocasionalmente ocurre tras una factura sea. Este tipo de metaplasia se contempla menos claramente como una respuesta de adap tacin. Mecanismos de metaplasia. La metaplasia no resulta de un cambio en el feno tipo de un tipo de clula diferenciada; en vez de ello, es el resultado de una rep rogramacin de las clulas madre que se sabe que existen en los tejidos normales, o de las clulas mesenquimales indiferenciadas presentes en el tejido conectivo. En un cambio metaplsico, estas clulas precursoras se diferencian a lo largo de una nu eva va. La diferenciacin de clulas madre hacia un linaje particular se lleva a cabo mediante seales generadas por citocinas, factores de crecimiento, y componentes de la matriz extracelular en el entorno de las clulas. En el proceso estn implicad os genes de diferenciacin y especficos de tejido, y se est identificando un nmero cr eciente de ellos14. Por ejemplo, las protenas morfogenticas seas, miembros de la su perfamilia TGF-p\ inducen la expresin condrognica u osteognica en las clulas madre a la vez que suprimen la diferenciacin hacia msculo o grasa15. Estos factores de cr ecimiento, que actan como desencadenantes externos, inducen a continuacin factores especficos de transcripcin que dirigen la cascada de genes especficos de fenotipo hacia una clula completamente diferenciada. En la mayora de los casos no est claro cmo se descontrolan estas vas normales para producir metaplasia. En el caso de def iciencia o exceso de vitamina A, se sabe que el cido retinoico regula el crecimie nto celular, la diferenciacin y los patrones tisulares y, de esta manera, puede i nfluir en la va de diferenciacin de las clulas madre16. Ciertos frmacos citostticos c ausan una alteracin de los patrones de metilacin del DNA y pueden transformar las clulas mesenquimales de un tipo (fibroblasto) a otro (msculo, cartlago). ESTIMULO AGRESOR 11 LESIN CELULAR REVERSIBLE Punto de irreversibilidad Estadio reversible? NECROSIS APOPTOSIS __ FIGURA 1-7 Estadios en la evolucin de la lesin y muerte celulares. Aspectos generales de la lesin y muerte celulares Como se dijo al principio del captulo, la lesin celular es el resultado de un estrs celular tan intenso que las clulas ya no son capaces de adaptarse o de una expos icin celular a agentes inherentemente lesivos. La lesin puede progresar a travs de un estadio reversible y culminar con la muerte celular (Fig. 1-7). En la Figura 1-8 se muestra una panormica de los cambios morfolgicos en la lesin celular. Las al teraciones bioqumicas y las anomalas morfolgicas asociadas se describen despus en el apartado de Mecanismos de lesin celular. Estas alteraciones pueden dividirse en lo s siguientes estadios: Las clulas irreversiblemente lesionadas sufren, invariablemente cambios morfolgico s que se reconocen como muerte celular. Exist dos tipos de muerte celular, la ne crosis y la apoptosis, que difieren en su morfologa, mecanismos y papeles en la e nfermedad y la fisiologa (Fig. 1-9 y Tabla 1-2). Cuando el dao a las membranas es intenso, las enzimas lisosomales penetran en el citoplasma y digieren la clula, y los contenidos celulares se derraman, dando lugar a necrosis. Algunos estmulos n ocivos, especialmente los que daan al DNA, inducen otro tipo de muerte, la apopto sis, que se caracteriza por disolucin nuclear sin prdida completa de la integridad de la membrana. Mientras que la necrosis siempre es un proceso patolgico, la apo ptosis sirve a muchas funciones normales y n se asocia necesariamente a lesin cel
ular. Aunque subrayamos las distinciones entre necrosis y apoptosis, puede haber algn solapamiento y mecanismos comunes entre estas dos vas. Adems, al menos alguno s tipos de estmulo pueden inducir apoptosis o necrosis, dependiendo de la intensi dad y duracin del estmulo, la rapidez del proceso de muerte y los trastornos bioqum icos inducidos en la clula lesionada. Los mecanismos y significado de estas dos va s de muerte se abordan despus en el captulo. En las siguientes secciones tratamos las causas y mecanismos de lesin celular. Primeramente, describimos las secuencia s de acontecimientos en la lesin celular y su punto final, la necrosis, y comenta mos ejemplos ilustrativos seleccionados de lesin y necrosis celular. Concluimos c on la discusin del patrn singular de la muerte celular representado por la apoptos is. Causas de lesin celular Lesin celular reversible. Inicialmente, la lesin se manifiesta Las causas de lesin celular van desde la violencia fsica grosera como cambios funcionales y morfolgico s reversibles si se elimiexterna de un accidente de automvil hasta causas endgenas na el estmulo daino. Las caractersticas de la lesin reversible internas, tales como una mutacin gentica sutil que produce la son la reduccin de la fosforilacin oxidati va, la deplecin del falta de una enzima vital que altera la funcin metablica normal . adenosintrifosfato (ATP), y la hinchazn celular producida por La mayora de los e stmulos lesivos pueden agruparse en las cambios en las concentraciones inicas y el aflujo de agua. siguientes categoras amplias. Lesin irreversible y muerte celular . Con el dao continuado, la Privacin de oxgeno. La hipoxia es una deficiencia de oxg elesin se hace irreversible, en cuyo momento la clula no puede no, que produce les in celular reduciendo la respiracin aerbirecuperarse. Hay un acontecimiento bioqumico crtico (el ca oxidativa. La hipoxia es una causa extremadamente imgolpe letal) res ponsable del punto de no retorno? No hay resportante y frecuente de lesin y muert e de la clula. Debe puestas claras a esta cuestin. Sin embargo, como se comenta di stinguirse de la isquemia, que es una prdida del riego sangudespus, en los tejidos isqumicos como el miocardio ciertos neo por un flujo sanguneo obstaculizado o por drenaje venoso cambios estructurales (p. ej., densidades amorfas mitocondriaredu cido en un tejido. La isquemia compromete el suministro les, indicativas de dao m itocondrial grave) y los cambios funno solamente de oxgeno, sino tambin de sustrat os metabbcionales (p. ej., prdida de permeabilidad de membrana) son cos, incluyend o glucosa (normalmente suministrada por el indicativos de clulas que han sufrido una lesin irreversible. riego sanguneo). Por lo tanto, los tejidos isqumicos se les ionan
12 UNIDAD I Patologa general rr o z ocr UJ w OLu Tumefaccin del retculo endoplsmico y mitocondrias Agrupamiento de grumos de cromatina Recuperacin Fragmentacin de la membrana celular y el ncleo ce rr o < L U UJ 55 O I O C Q LUCE - I LU Tumefaccin del retculo endoplsmico y prdida de ribosomas Rotura lisosomal Protrusion es de la membrana Figuras de mielina Necrosis Condensacin nuclear Mitocondrias hinchadas con densidades amorfas w > (:. > VA J / UJ rr FIGURA 1-8 Representacin esquemtica de una clula normal y los cambios en la lesin ce lular reversible e irreversible. Se muestran cambios morfolgicos, que se describe n en las pginas siguientes y se muestran en microfotografas electrnicas en la Figur a 1-17. La agresin reversible se caracteriza por tumefaccin generalizada de la clul a y sus organelas; protrusiones en la membrana plasmtica; desprendimiento de los ribosomas del retculo endoplsmico, y agrupacin en grumos de la cromatina nuclear. L a transicin a lesin irreversible se caracteriza por un aumento de la tumefaccin de la clula, hinchazn y rotura de los lisosomas; presencia de grandes densidades amor fas en las mitocondrias hinchadas; rotura de las membranas celulares, y cambios nucleares profundos. Los ltimos incluyen condensacin nuclear (picnosis), seguida d e fragmentacin (cariorrexis) y disolucin del ncleo (carilisis). Las estructuras lami nares (figuras de mielina) derivadas de las membranas daadas de las organelas y d e la membrana plasmtica aparecen primero durante el estadio reversible y se hacen ms pronunciadas en las clulas daadas irreversiblemente. Los mecanismos subyacentes
a estos cambios se comentan en el texto que sigue. ms rpido y ms intensamente que los tejidos hipxicos. Una causa de hipoxia es la oxig enacin inadecuada de la sangre debida a una insuficiencia cardiorrespiratoria. La prdida de la capacidad de transporte de oxgeno por la sangre, como en la anemia o en el envenenamiento por monxido de carbono (que produce una monoxihemoglobina e stable que bloquea el transporte de oxgeno), es una causa menos frecuente de priv acin de oxgeno que resulta en una lesin significativa. Dependiendo de la gravedad d el estado hipxico, las clulas pueden adaptarse, sufrir lesin o morir. Por ejemplo, si la arteria femoral se estrecha, las clulas musculares esquelticas de la pierna pueden disminuir de tamao (atrofia). Esta reduccin en la masa celular consigue un equilibrio entre las necesidades metablicas y el suministro disponible de oxgeno. Una hipoxia ms intensa induce lesin y muerte celular. Agentes fsicos. Los agentes fs icos capaces de producir lesin celular incluyen el traumatismo mecnico, las temper aturas extremas (quemaduras y fro intenso), los cambios sbitos en la presin atmosfrica, la radia cin y la descarga elctrica (Captulo 9). Agentes qumicos y frmacos. La lista de agente s qumicos que pueden producir lesin celular desafa su compilacin. Los productos qumic os simples tales como la glucosa o la sal en concentraciones hipertnicas pueden p roducir lesin celular directamente o alterando la homeostasia electroltica de las clulas. Incluso el oxgeno, en concentraciones altas, es altamente txico. Cantidades mnimas de agentes conocidos como venenos, tales como arsnico, cianuro o sales de mercurio, pueden destruir el suficiente nmero de clulas en minutos u horas para pr oducir la muerte. Otras sustancias, sin embargo, son nuestros compaeros diarios: contaminantes ambientales y areos, insecticidas y herbicidas; riesgos industriale s y ocupacionales, como el monxido de carbono y el asbestos; estmulos sociales, ta les como alcohol y narcticos, y la variedad siempre creciente de drogas teraputica s.
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 13 NORMAL FIGURA 1-9 Cambios ultraestructurales secuenciales vistos en la necrosis (izquie rda) y la apoptosis (derecha). En la apoptosis, los cambios iniciales consisten en condensacin de la cromatina nuclear y fragmentacin de la misma, seguidas de for macin de protrusiones citoplasmticas y fagocitosis de los cuerpos apoptticos formad os. Signos de protrusiones citoplasmticas y digestin y escape de los componentes c elulares. (Adaptado de Walker NI, et al: Patterns of cell death. Methods Archiv Exp Pathol 13:18-32, 1988. Reproducido con permiso de S. Karger AG, Basiel.) Digestin enzimtica y escape de los contenidos celulares _ 't& ' >fSj| Fagocito NECROSIS APOPTOSIS Fagocitosis de clulas apoptticas y fragmentos TABLA 1-2 Caracterstica Tamao celular Ncleo Membrana plasmtica Contenidos celulares Inflamacin adyacente Papel fisiolgico o patolgico Necrosis Agrandado (tumefaccin) Caractersticas de necrosis y apoptosis Apoptosis Reducido (encogimiento) Desinteg racin en fragmentos de tamao de nucleosoma Intacta; estructura alterada, especialm ente la orientacin de los lpidos Intactos; pueden liberarse en cuerpos apoptticos N o A menudo fisiolgica, forma de eliminar clulas no deseadas; puede ser patolgica tr as algunas formas de agresin celular, especialmente el dao al DNA Picnosis - cariorrexis - carilisis Rota : Digestin enzimtica; pueden salir de la clula Frecuente Invariablemente patolgico (culminacin de lesin celular irreversible) Agentes infecciosos. Los agentes abarcan desde virus submicroscpicos hasta las gr andes tenias. En medio estn las rickettsias, bacterias, hongos y las formas ms evo lucionadas de parsitos. Las maneras por las que este grupo heterogneo de agentes b iolgicos producen lesin son diversas y se abordan en el Captulo 8. Reacciones inmun olgicas. Aunque el sistema inmunitario sirve como funcin esencial en la defensa co ntra patgenos infecciosos, las reacciones inmunitarias, de hecho, pueden producir lesin celular. La reaccin anafilctica ante una protena extraa o un frmaco es un buen ejemplo, y las reacciones ante autoantgenos endgenos son responsables de varias en fermedades autoinmunitarias (Captulo 6). Trastornos genticos. Los defectos genticos como causas de lesin celular son del mayor inters para cientficos y mdicos hoy da (C aptulo 5). La lesin gentica puede dar lugar a un defecto tan grave como las malform aciones congnitas asociadas con el sndrome de Down, producido por una anomala cromo smica, o tan sutil como la disminucin en la vida de los hemates producida por una s ustitucin de un nico aminocido en la hemoglobina S en la anemia falciforme. Los div ersos errores innatos del metabolismo derivados de anomalas enzimticas, usualmente la falta de una enzima, son excelentes ejemplos de dao celular debido a alteraciones sutiles en el DNA. Las variaciones en la dotacin gentica pueden influir tambin sob re la susceptibilidad de las clulas a la lesin por productos qumicos y otras agresi ones ambientales. Desequilibrios nutricionales. Los desequilibrios nutricionales siguen siendo causas importantes de lesin celular. Las deficiencias proteinocalri cas producen un nmero alarmante de muertes, fundamentalmente en las poblaciones m enos privilegiadas. Las deficiencias de vitaminas especficas se encuentran en tod o el mundo (Captulo 9). Los problemas nutricionales pueden ser autoimpuestos, com o ocurre en la anorexia nerviosa o en la inanicin autoinducida. Irnicamente, los e
xcesos nutricionales tambin han llegado a ser causas importantes de lesin celular. Los excesos de lpidos predisponen a la aterosclerosis, y la obesidad es una mani festacin de sobrecarga grasa de algunas clulas corporales. La aterosclerosis es vi rtualmente endmica en Estados Unidos, y la obesidad sigue aumentando. Adems de los problemas de hiponutricin e hipernutricin, la composicin de la dieta contribuye si gnificativamente a varias enfermedades. Las enfermedades metablicas tales como la diabetes tambin causan una lesin celular grave.
14 UNIDAD I Patologa general Mecanismos de lesin celular Los mecanismos bioqumicos responsables de la lesin celular son complejos. Sin emba rgo, hay un nmero de principios que tienen que ver con la mayora de las formas de lesin celular: DEPLECION DE ATP La deplecin de ATP y la sntesis disminuida de ATP se asocian frecuentemente con le sin hipxica y qumica (txica) (Fig. 1-11). Se requiere fosfato de alta energa en forma de ATP para muchos procesos sintticos y degradativos en la clula. stos incluyen el La respuesta celular a los estmulos lesivos depende del tipo transporte de membr ana, la sntesis de protena, la lipognesis y de lesin, su duracin y su intensidad. As, las dosis pequeas de las reacciones de desacetilacin-reacetilacin necesarias para e l ciclo fosfolipdico. El ATP se produce de dos maneras. La va ms una toxina qumica o breves perodos de isquemia pueden importante en las clulas del mamfero es la fosfo rilacin oxidatiinducir una lesin reversible, mientras que las dosis grandes de va del adenosindifosfato, en una reaccin que da lugar a la reducla misma toxina o un a isquemia ms prolongada podran dar cin del oxgeno mediante el sistema de transferen cia de electrolugar a la muerte celular instantnea o una lesin lenta irrevernes en las mitocondrias. La segunda es la va glucoltica, que puede sible dando lugar a l a muerte celular con el tiempo. producir ATP en ausencia de oxgeno utilizando glu cosa derivada Las consecuencias de la lesin celular dependen del tipo, estado y a daptabilidad de la clula lesionada. El estado nutricional y hor-de los lquidos cor porales o de la hidrlisis de glucgeno. As, los monal de la clula y sus necesidades m etablicas son importantes en la respuesta a la lesin. Cuan vulnerable es, por ejemp lo, Isquemia una clula ante la prdida del riego sanguneo e hipoxia? La clula muscula r estriada en la pierna puede colocarse enteramente en reposo cuando se le priva de su riego sanguneo, no as el msculo estriado del corazn. La exposicin de dos indiv iduos a concentraciones idnticas de una toxina, como el tetracloruro de carbono, puede no producir el efecto en uno y muerte celular en Mitocondria el otro. Esto puede deberse a variaciones genticas que afectan a la cantidad y actividad de en zimas hepticas que convierten el tetracloruro de carbono en productos intermedios txicos I Fosforilacin oxidativa (Captulo 9). Con el mapa del genoma humano complet o existe un gran inters en la identificacin de polimorfismos genticos que afecten a la respuesta celular ante agentes lesivos. tATP La lesin celular es el resultado de anomalas funcionales y bioqumicas en uno o ms de los varios componentes celular es esen- i Bomba de Na+ t Gluclisis anaerbica Otros ciales (Fig. 1-10). Las dianas ms importantes de los estmulos I efectos lesivos son: (1) la respiracin aerbica que implica la fosforilacin oxidativa mitocondrial y la produccin de ATP; (2) la t En trada de Ca ++ * Glucgeno tpH Desprendimiento integridad de las membranas celular es, de la cual dependen H2O y Na+ de ribosomas, etc. la homeostasia inica y osmtic a de la clula y sus organelas; (3) la sntesis proteica; (4) el citoesqueleto, y (5 ) la integridad t Salida de K+ { j del aparato gentico de la clula. Grumos * Sntesi s 1 J f ^ 1 1
En la seccin siguiente describimos cada uno de los mecanismos bioqumicos responsab les de la lesin celular inducida por diferentes estmulos. Debe notarse que con la mayora de los estmulos mltiples mecanismos contribuyen a la lesin y, en el caso de m uchos estmulos lesivos, la localizacin bioqumica real de la lesin permanece desconoc ida. de cromatina proteica Tumefaccin del RE nuclear I Tumefaccin celular " Prdida Depsit o de microvellosidades de lpidos Protrusiones FIGURA 1-11 Consecuencias funcional es y morfolgicas de la disminucin intracelular de ATP durante la agresin celular. ESTMULO AGRESOR \ ATP DAO DE MEMBRANA Mitocondria Lisosoma Membrana plasmtica f Ca 2+ INTRACELULAR I FORMAS DE OXGENO REACTIVO o2H202 OH* <0> Prdida de funciones celulares dependientes de energa Muerte celular FIGURA 1-10 \ Digestin Prdida enzimtica de contenidos de componentes de la clula celulares Fragmen tacin proteica Dao al DNA Sitios celulares y bioqumicos de dao en la agresin celular.
CAPTULO ( y Adaptaciones celulares, lesin celular y muerte celular 15 alta conductibilidad, lo que se ha llamado transicin a la permeabilidad mitocondr ial, en la membrana interna de la mitocondria (Fig. 1-12)17. Aunque es reversibl e en los primeros estadios, este poro no selectivo se hace permanente si persist e el estmulo desencadenante, impidiendo el mantenimiento de la fuerza protnica mit ocondrial, o potencial. Dado que el mantenimiento del La actividad de la bomba d e sodio de la membrana plasmti- potencial de membrana es crtico para la fosforilac in oxidativa ca dependiente de energa (ATPasa-Na+,K+ sensible a ouabana) mitocondri al, se desprende que la transicin a la permeabilidad est reducida. La insuficienci a de este sistema de transporte mitocondrial irreversible es un golpe mortal par a la clula. El dao mitocondrial tambin puede asociarse con salida del citoactivo, d ebido a la concentracin disminuida de ATP y aumencromo c al citosol. Dado que el citocromo c es un componente to de la actividad ATPasa, da lugar a la acumulacin intraceluintegral de la cadena de transporte de electrones y puede desenlar de s odio y a que el potasio salga de la clula. La ganancia cadenar las vas de la muert e apopttica en el citosol (ver ms neta de solutos se acompaa por ganancia isosmtica de agua, produciendo tumefaccin celulary dilatacin del retculo endo- adelante), est e acontecimiento patolgico es, asimismo, con probabilidad un determinante clave d e la muerte celular. plsmico (verFig. 1-8). El metabolismo energtico celular est al terado. Si el suministro de oxgeno a las clulas se reduce, como ocurre en la isque mia, la fosforilacin oxidativa cesa y las clulas dependen de la Lesin o disfuncin mi tocondrial gluclisis para la produccin de energa. Este cambio al meta(aumento de Ca 2+ citoslico, estrs oxidativo, peroxidacin lipdica) bolismo anaerbico est controlado por metabolitos de la va de energa que actan sobre enzimas glucolticas. La disminucin en ATP celular y el aumento asociado en adenosn monofosfato estimulan las activi dades de fosfofructocinasa y fosforilasa. Esto da lugar a una tasa aumentada de gluclisis anaerbica diseada para mantener la fuente de energa celular mediante la ge neracin de ATP por el metabolismo de la glucosa derivada del glucgeno. Como consec uencia, los almacenes de glucgeno se agotan rpidamente. La gluclisis tiene como res ultado la acumulacin de cido lctico y fosfatos inorgnicos de la hidrlisis de los este res de fosfato. Esto reduce el pH intracelular dando lugar a disminucin de la act ividad de muchas enzimas celulares. El fallo de la bomba de Ca2+ da lugar a entr ada de Ca2+, con efectos lesivos sobre numerosos componentes celulares, descrito s ms adelante. Con la deplecin prolongada o empeorada de ATP ocurre una alteracin e structural del aparato sinttico de protenas, Transicin otras protenas manifestada co mo un desprendimiento de ribosomas del rede la permeabilidad pro-apoptticas tculo endoplsmico rugoso y una disociacin de polisomas a mitocondrial (TPM) monosomas, c on una reduccin consecuente en la sntesis proteica. Alfinal,hay un dao irreversible en las mitocondrias y las Apoptosis membranas lisosomales, y la clula sufre necr osis (ver Fig. 1 -8). FIGURA 1-12 Disfuncin mitocondrial en la lesin celular. En l as clulas privadas de oxgeno o glucosa, las protenas pueden plegarse errneamente, y estas protenas mal plegadas desencadenan una reaccin celular denominada respuesta de la protena desplegada que puede dar lugar a lesin celular e inclu- AFLUJO DE CA LCIO IIMTRACELULAR so muerte. Este proceso se describe ms adelante en el captulo. Y PRDIDA DE LA HOMEOSTASIA El plegamiento defectuoso de la protena tambin se ve en las DEL CALCIO clulas expuestas a estrs, tales como calor, y cuando las proLos ion es de calcio son mediadores importantes de la lesin celutenas se daan por enzimas ( tales como enzimas que responlar18. El calcio citoslico libre se mantiene a conce ntraciones den a Ca2+, descritas ms adelante) y radicales libres. extremadamente bajas (siendo menor de 0,1 umol) en comparacin con los niveles extracelulares de 1,3 mmol, y la mayora del DAO MITOCONDRIAL calcio intracelular est secuestrado en l as mitocondrias y en el Las mitocondrias son dianas importantes de virtualmente todos retculo endoplsmico. Tales gradientes estn modulados por los tipos de estmulos lesivos, incluyendo hipoxia y toxinas. FreATPasas-Mg2+,Ca2+, dependientes de en erga, asociadas a la memcuentemente, el dao celular se acompaa de cambios morfolgibr
ana. La isquemia y ciertas toxinas producen un aumento precoz cos en las mitocon drias. Las mitocondrias pueden lesionarse por en la concentracin citoslica de calc io, debido al aflujo neto de aumentos del Ca2+ citoslico, por estrs oxidativo, por degradaCa2+ a travs de la membrana plasmtica y la liberacin de Ca2+ de cin de fosfo lpidos a travs de las vas de fosfolipasa A2 y esfingo- las mitocondrias y del retcul o endoplsmico (Fig. 1-13). Los mielina, y por productos de la degradacin de los lpi dos derivaaumentos mantenidos subsiguientes de Ca2+ intracelular derivan dos de aqu, tales como cidos grasos libres y ceramida. A de aumentos inespecficos en la pe rmeabilidad de membrana. menudo, el dao mitocondrial da lugar a la formacin de can al de A su vez, el aumento de Ca2+ activa varias enzimas, con efectos tejidos con mayor capacidad glucoltica (p. ej., hgado) estn en ventaja cuando dismi nuyan los niveles de ATP por la inhibicin del metabolismo oxidativo por la lesin. La deplecin del ATP a menos del 5 al 10% de los niveles normales tiene efectos am plios sobre muchos sistemas celulares crticos:
16 UNIDAD I Patologa general Los radicales libres son especies qumicas que tienen un nico electrn sin su par en una rbita externa. La energa creada por esta configuracin inestable se libera a tra vs de reacciones con molculas adyacentes, tales como productos qumicos inorgnicos u orgnicos -protenas, lpidos, hidratos de carbono- particularmente con molculas clave en las membranas y cidos nucleicos. Adems, los radicales libres inician reacciones autocatalticas, en las cuales las molculas con las que reaccionan se convierten a su vez en radicales libres, propagando el dao en cadena. Los radicales libres pu eden iniciarse dentro de las clulas de diversas maneras (Fig. 1-14): FIGURA 1-13 Fuentes y consecuencias del aumento de calcio citoslico en la agresin celular. ATP: adenosintrifosfato. celulares potencialmente deletreos. Las enzimas que se sabe que se activan por el calcio incluyen ATPasas (que por lo tanto aceleran la deplecin del ATP), fosfoli pasas (que producen lesin en la membrana), proteasas (que rompen las protenas de m embrana y citoesquelticas), y endonucleasas (que son responsables de la fragmenta cin del DNA y la cromatina). Los niveles aumentados de Ca2+ intracelular tambin da n lugar a un aumento de la permeabilidad mitocondrial e induccin de apoptosis18"2 0. Aunque la lesin celular a menudo origina un aumento de calcio intracelular y, a su vez, esto media varios efectos deletreos, incluyendo muerte celular, la prdid a de la homeostasia de calcio no siempre es un episodio proximal en la lesin celu lar irreversible. ACUMULACIN DE RADICALES LIBRES DERIVADOS DE OXGENO (ESTRS OXIDATI VO) Las clulas generan energa reduciendo el oxgeno molecular a agua. Durante este proce so se producen pequeas cantidades de Los efectos de estas especies reactivas son de un rango amplio, formas reactivas de oxgeno parcialmente reducidas como un per o tres reacciones son particularmente relevantes en la lesin subproducto inevitab le de la respiracin mitocondrial. Algunas celular (ver Fig. 1-14): de estas forma s son radicales libres que pueden daar lpidos, protenas y cidos nucleicos. Son refer idos como especies de oxge Peroxidacin lipdica de membranas. Los radicales libres en no reactivo. Las clulas tienen sistemas de defensa para prevenir la presencia de oxgeno pueden producir peroxidacin de lpidos dentro del plasma y de las membranas de las organelas. El dao lesin causada por estos productos. Un desequilibrio entre los sisoxidativo se inicia cuando los enlaces dobles de los cidos gratemas gener adores y limpiadores de radicales libres da lugar a sos insaturados de los lpidos de la membrana son atacados por estrs oxidativo, una situacin que se ha asociado con la lesin radicales libres derivados del oxgeno, particularmente por OH. celula r observada en muchas afecciones patolgicas. El dao Las interacciones lpido-radical libre dan lugar a perxidos, que mediado por radicales libres contribuye a proces os diversos como a su vez son inestables y reactivos, y se siguen de una reaccin en la lesin qumica y por radiacin, la lesin de isquemia-reperfucadena autocataltica ( denominada propagacin), que puede sin (inducida por la restauracin del riego sangune o en el tejidar lugar a un dao extenso en la membrana, organelas y en la do isqumi co), el envejecimiento celular y la muerte microbiana clula. Otras opciones de te rminacin ms favorables tienen por los fagocitos21"24. Absorcin de la energa radiante (p. ej., luz ultravioleta, rayos X). Por ejemplo, l a radiacin ionizante puede hidrolizar el agua en radicales libres hidroxilo (OH) e hidrgeno (H). Metabolismo enzimtico de agentes qumicos o frmacos exgenos (p. ej., t etracloruro de carbono [CCLJ que puede generar CC13, descrito ms adelante en este captulo). Las reacciones de reduccin-oxidacin que ocurren durante los procesos met ablicos normales. Durante la respiracin normal, el oxgeno molecular se reduce secue ncialmente mediante la adicin de cuatro electrones para generar agua. Tal convers in tiene lugar mediante enzimas oxidativas en el retculo endoplsmico, citosol, mito condrias, peroxisomas y lisosomas. En este proceso se producen pequeas cantidades
de intermediarios txicos; stos incluyen el radical anin superxido (CV), perxido de h idrgeno (H202) e iones hidroxilo (OH). Durante la inflamacin, ocurren estallidos rp idos de produccin de superxido en los leucocitos polimorfonucleares activados. Est o ocurre por una reaccin muy precisamente controlada en un complejo multiprotena d e la membrana plasmtica que utiliza NADPH oxidasa para la reaccin rdox (Captulo 2). Adems, algunas oxidasas intracelulares (como la xantina oxidasa) generan radicale s superxido como consecuencia de su actividad. Metales de transicin tales como hie rro y cobre donan o aceptan electrones libres durante las reacciones intracelula res y catalizan la formacin de radicales libres, como en la reaccin de Fenton (H20 2 + Fe2+ -+ Fe3+ + OH + OH"). Como la mayora del hierro libre intracelular est en estado frrico (Fe3+), debe reducirse primeramente a la forma ferrosa (Fe2+) para participar en la reaccin de Fenton. Esta reduccin puede potenciarse por superxido, y por ello se requieren fuentes de hierro y superxido para un dao celular oxidativ o mximo. El xido ntrico (ON), un mediador qumico importante generado por las clulas e ndoteliales, macrfagos, neuronas y otros tipos celulares (Captulo 2), puede actuar como radical libre y tambin puede convertirse en un anin peroxinitrito altamente reactivo (ONOO~) as como en N0 2 y N03".
CAPTULO 1 i A. PRODUCCIN DE RADICALES LIBRES Adaptaci ones celulares, lesin celular y muerte celular 17 Inflamacin Radiacin Toxicidad del oxgeno Agentes qumicos Lesin por reperfusin Oxidasa P450 Enzimas de la cadena respiratoria Enzimas citoslicas Oxidasa NADPH' Oxidasa Formas de oxgeno reactivo: 0 2 T , H 2 0 2 , OH Formas de oxgeno reactivo 0 2 T , H 2 0 2 , OH" Todas las membranas Vitaminas E y A p-caroteno Fe+2 Fenton Fe+3 . SOD Og* > H2O2 -+=*:OH-+OHCatalasa H Peroxidacin de los lpidos de membrana Glutatin peroxidasa GSSG \ 2GSH Glutatin reductasa \ H20 2 Fragmentacin del DNA Entrecruzamiento y fragmentacin proteicos Citosol SOD Vitamina C Glutatin peroxidasa Ferritina Ceruloplasmina Mitocondria SOD Glutatin peroxidasa Peroxisomas Catalasa B. LESIN CELULAR POR RADICALES LIBRES C. NEUTRALIZACIN DE RADICALES LIBRES (AUSENCIA DE LESIN CELULAR) FIGURA 1-14 Papel de las formas de oxgeno reactivo en la lesin celular. El 0 2 se convierte en superxido (02") por enzimas oxidativas en el retculo endoplsmico (RE), mitocondrias, membrana plasmtica, peroxisomas y citosol. El 0 2 " se convierte e n H202 por dismutacin y, de ah, a OH por la reaccin de Fenton catalizada por Cu27Fe 2+. El H202 tambin se deriva directamente de las oxidasas de los peroxisomas. No se muestra otro radical potencialmente lesivo, el oxgeno singlet. Los radicales l ibres resultantes daan a los lpidos (peroxidacin), a las protenas y al DNA, lo que c onduce a diversas formas de lesin celular. Ntese que el superxido cataliza la reduc cin de Fe3+ a Fe2*, aumentando as la produccin de OH por la reaccin de Fenton. Las p rincipales enzimas antioxidantes son las superxido dismutasa (SOD), la catalasa y
la glutatin peroxidasa. GSH: glutatin reducido; GSSG: glutatin oxidado; NADPH: for ma reducida de la nicotinamida adenina dinucletido fosfato. lugar cuando el radical libre se captura por un limpiador tal como la vitamina E , embutida en la membrana celular. Modificacin oxidativa de protenas. Los radicale s libres favorecen la oxidacin de las cadenas laterales de los residuos de aminoci do, formacin de enlaces cruzados protena-protena (p. ej., enlaces disulfuro), y oxi dacin del esqueleto proteico, dando lugar a la fragmentacin proteica. La modificac in oxidativa potencia la degradacin de protenas crticas mediante el complejo multica taltico proteosoma, causando estragos en la clula. Lesiones en el DNA. Las reaccio nes con la timina en el DNA nuclear y mitocondrial producen roturas de una caden a del DNA. Este dao en el DNA se ha implicado en el envejecimiento celular (comen tado ms adelante en este captulo) y en la transformacin neoplsica de las clulas (Captu lo 7). Las clulas han desarrollado mltiples mecanismos para eliminar los radicales libres y, por lo tanto, para minimizar la lesin. Los radicales libres son inhere ntemente inestables y, en general, se degradan espontneamente. Por ejemplo, el su perxido es inestable y se degrada (se dismuta) espontneamente en oxgeno y agua oxig enada en presencia de agua. Sin embargo, existen varios sistemas no enzimticos y enzimticos que contribuyen a la inactivacin de las reacciones de radic ales libres (ver Fig. 1-14). Son los siguientes: Los antioxidantes o bien bloque an el inicio de la formacin del radical libre o lo inactivan (p. ej., eliminan) r adicales libres y terminan el dao radical. Son ejemplos las vitaminas liposoluble s E y A as como el cido ascrbico y el glutatin en el citosol. Segn hemos visto, el hi erro y el cobre pueden catalizar la formacin de especies de oxgeno reactivo. Los n iveles de estas formas reactivas se minimizan mediante la unin de iones a las pro tenas de almacenamiento y de transporte (p. ej., transferrina, ferritina, lactofe rrina y ceruloplasmina), minimizando con ello la formacin de OH. Una serie de enz imas acta como sistemas de eliminacin de radicales libres y descompone el perxido d e hidrgeno y el anin superxido. Estas enzimas se localizan cerca de los sitios de g eneracin de estos oxidantes e incluyen las siguientes: Catalasa, presente en pero xisomas, que descompone el H 2 0 2 (2 H 2 0 2 ^ 0 2 + 2 H 2 0). Superxido dismuta sas, que se encuentran en muchos tipos celulares y convierten el superxido en H 2 0 2 (2 02~+ 2 H >
18 UNIDAD I % Patologa general H202 + 02)23. Este grupo incluye la manganeso-superxid o dismutasa, que se localiza en la mitocondria, y la cobrecinc-superxido dismutas a, que se encuentra en el citosol. Glutatin peroxidasa, que protege tambin contra la lesin al catalizar la descomposicin de radicales libres (H 2 0 2 + 2 GSH - GSSG [homodmero glutatin] + 2 H 2 0, o 2 OH + 2 GSH - GSSG + 2 H 2 0). La proporcin intr acelular de glutatin oxidado (GSSG) respecto a glutatin reducido (GSH) es un refle jo del estado oxidativo de la clula, y es un aspecto importante de la capacidad c elular para desintoxicar especies de oxgeno reactivo. a todas las membranas celul ares, incluyendo las mismas mitocondrias. A la vez, el aumento en el calcio cito slico, asociado con deplecin del ATP, da lugar a una captacin aumentada de Ca2+ por las mitocondrias, activando las fosfolipasas y dando lugar a una descomposicin d e los fosfolpidos. El resultado neto es la deplecin de fosfolpidos de las mitocondr ias y otras membranas celulares, y la acumulacin de cidos grasos libres. En la mit ocondria, estos cambios producen defectos de permeabilidad, tales como los de tr ansicin de la permeabilidad de la mitocondria17,26 dando lugar a una lesin celular progresiva (ver Fig. 1-12). Prdida de fosfolpidos de membrana. La lesin celular gr ave se asocia con una disminucin en el contenido de fosfolpidos de membrana, por l a degradacin debida probablemente a la activacin de fosfolipasas endgenas por los n iveles aumentados de calcio citoslico26. La prdida fosfolipdica tambin puede ocurrir secundariamente a una disminucin de la reacetilacin dependiente de ATP o a una di sminucin de la sntesis de novo de fosfolpidos. Anormalidades citoesquelticas. Los fi lamentos citoesquelticos sirven como anclajes que conectan la membrana plasmtica c on el interior de la clula. La activacin de proteasas por el aumento del calcio ci toslico puede producir dao en los elementos del citoesqueleto. En presencia de tum efaccin celular, este cambio da lugar, particularmente en las clulas miocrdicas, al despegamiento de la membrana celular del citoesqueleto haciendo que sta sea susc eptible a la distensin y rotura. Especies de oxgeno reactivo. Los radicales libres de oxgeno parcialmente reducido causan lesin en las membranas celulares y en otro s constituyentes de la clula mediante mecanismos que se discutieron antes. Produc tos de descomposicin de los lpidos. Incluyen los cidos grasos libres no esterificad os, la acilcarnitina y los lisofosfolpidos, productos catablicos que se sabe que s e acumulan en las clulas lesionadas como resultado de la degradacin fosfolipdica. T ienen un efecto detergente sobre las membranas. Asimismo, pueden bien insertarse en la bicapa lipdica de la membrana o bien intercambiarse con los fosfolpidos de memEn muchos procesos patolgicos, los efectos finales inducidos por los radicales li bres dependen del equilibrio neto entre la formacin de radical libre y su termina cin. Como se ha afirmado antes, se piensa que los radicales libres estn implicados en muchos procesos patolgicos yfisiolgicos,que se mencionarn a lo largo del libro. DEFECTOS EN LA PERMEABILIDAD DE MEMBRANA La prdida precoz de la permeabilidad selectiva de la membrana, que conducefinalme ntea un dao manifiesto de la misma, es una caracterstica constante de la mayora de las formas de lesin celular. El dao de la membrana puede afectar a las mitocondria s, la membrana plasmtica y otras membranas celulares. En las clulas isqumicas, los defectos de membrana pueden ser el resultado de una serie de acontecimientos que implican la deplecin del ATP y la activacin de las fosfolipasas moduladas por cal cio (ver ms adelante). Sin embargo, la membrana plasmtica puede daarse tambin direct amente por ciertas toxinas bacterianas, protenas vricas, componentes lticos del com plemento, y una variedad de agentes fsicos y qumicos. Varios mecanismos bioqumicos pueden contribuir al dao de la membrana (Fig. 1-15): Disfuncin mitocondrial. La fu ncin mitocondrial defectuosa da lugar a una sntesis disminuida de fosfolpido, que a fecta f Ca 2+ citoslico Disfuncin mitocondrial ^ Reacilacin/ sntesis de fosfolpidos
Activacin de fosfolipasa Activacin de proteasa f Degradacin de fosfolpidos Dao \ i . citoesqueltico^W^. 1 | \ FIGURA 1-15 Mecanismos del dao de la membrana en la lesin celular. La disminucin de 0 2 y el aumento del Ca2* citoslico se ven, tpicamente, en la isquemia pero puede n acompaar a otras formas de lesin celular. Las formas de oxgeno reactivo, que se p roducen a menudo en la reperfusin de los tejidos isqumicos, producen tambin dao de l a membrana (no mostrado). I Productos Prdida de la de fosfolpidos degradacin lipdica DAO DE MEMBRANA
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular 19 sin original. El segundo es el desarrollo de intensos trastornos en funcin de memb rana. Como se mencion antes, la agresin a las membranas hsosomales da lugar a libe racin de sus enzimas al El dao en las membranas mitocondriales tiene consecuencias citoplasma; las hidrolasas acidas se activan con la disminucin del que se descri bieron anteriormente. El dao en la membrana plaspH intracelular de una clula isqumi ca y degradan los compomtica da lugar a prdida del equilibrio osmtico y aflujo de n entes citoplsmicos y nucleares. Esta disolucin de la clula lquidos e iones, as como a una prdida de protenas, enzimas, lesionada es caracterstica de la necrosis, uno de los patrones recocoenzimas y cidos ribonucleicos. Las clulas tambin pueden nocidos de la muerte celular. Existe tambin una liberacin diseperder metabolitos, que son vitales en la reconstitucin del ATP, minada de enzimas celulares potencialmente destructivas al espavaciando as an ms la carga neta de fosfatos intracelulares de c io extracelular, con dao de los tejidos adyacentes y una alta energa. La lesin de l as membranas Hsosomales da lugar a larespuesta del husped (Captulo 2). Cualesquier a que sean los liberacin de sus enzimas en el citoplasma y activacin de las mismec anismos del dao de membrana, el resultadofinales una libemas. Los lisosomas conti enen RNasas, DNasas, proteasas, fosfata- racin masiva de materiales intracelulare s y un aflujo masivo de sas, glucosidasas y catepsinas. La activacin de esas enzi mas da calcio, con las consecuencias descritas anteriormente. lugar a digestin en zimtica de los componentes celulares, provoMerece la pena subrayar que la liberac in a la circulacin pericando la prdida de la ribonucleoprotena, desoxirribonucleofric a de protenas intracelulares a travs de la membrana celular protena y glucgeno, y la s clulas mueren por necrosis. degradada proporciona un medio de detectar la lesin y muerte celular especfica de tejido utilizando muestras de suero sanguneo. El mscu lo cardaco, por ejemplo, contiene una isoforma Lesin celular reversible especfica d e la enzima creatincinasa y de la protena contrctil e irreversible troponina; el hg ado (y especficamente el epitelio de las vas biliares) contiene una forma termorre sistente en la enzima fosfatasa Hasta este momento hemos enfocado nuestra atencin sobre las causas y mecanismos bioqumicos generales de la lesin celular. En alcali na, y los hepatocitos contienen transaminasas. La lesin irreversible y la muerte celular en estos tejidos se reflejan, en conseesta seccin volcaremos nuestra aten cin sobre las vas subyacentes cuencia, en niveles aumentados de tales protenas en l a sangre. a la secuencia de sucesos por los cuales la lesin reversible se hace ir reversible, dando lugar a la muerte celular, principalmente necrosis. Como se ha comentado previamente, los primeros cambios Morfologa de la lesin asociados con l as diversas formas de lesin celular son la producy necrosis celulares cin disminui da de ATP, la prdida de la integridad de la membrana celular, los defectos en la sntesis de protenas, el dao en el Las clulas sufren cambios secuenciales bioqumicos y morfolgicitoesqueleto y el dao en el DNA. Con ciertos lmites, la clula cos segn se l esionan progresivamente y, al final, mueren por puede compensar esos trastornos y, si disminuye el estmulo lesinecrosis. Todos los tipos de estrs e influencias no civas ejercen sus vo, volver a la normalidad. Sin embargo, la lesin persistente o efectos primero a un nivel molecular o bioqumico. Hay un hiato de excesiva hace q ue las clulas traspasen el umbral hasta la lesin tiempo entre el estrs y los cambio s morfolgicos de la lesin o irreversible (ver Fig. 1-8). Esto se asocia con un gra n dao en todas muerte celular; la duracin de este retraso puede variar de acuerdo las membranas celulares, hinchazn de lisosomas y vacuolizacin con la sensibilidad de los mtodos utilizados para detectar dichos de las mitocondrias con capacidad r educida para producir ATP. El cambios (Fig. 1-16). Con las tcnicas histoqumicas o ultraestruccalcio extracelular penetra en la clula y los almacenes de calcio tura les, los cambios pueden verse en minutos u horas tras la lesin intracelulares se vacan, dando lugar a la activacin de enzimas isqumica; sin embargo, puede tardarse considerablemente ms que pueden catabolizar membranas, protenas, ATP y cidos tiempo (horas a das) antes de que los cambios puedan verse con nucleicos. Tras esto, ex iste una prdida continua de protenas, coenzimas esenciales y cidos ribonucleicos a travs de la membrana plasmtica hiperpermeable, perdindose metabolitos celulares que
son vitales para la reconstitucin del ATP y agotando Cambios Lesin celular Lesin c elular an ms los fosfatos intracelulares de alta energa. Cambios al reversible irre versible Los mecanismos moleculares que conectan la mayora de las ultraestructura les microscopio formas de lesin celular con la muerte celularfinalse nos escapan p tico por varias razones. Primeramente, existen claramente muchas vas de lesin celu lar, no todas ellas invariablemente letales. En segundo lugar, las numerosas mac romolculas, enzimas y organelas dentro de la clula son tan estrechamente interdepe ndientes que es difcil distinguir una lesin primaria de efectos secundarios (y no necesariamente importantes). En tercer lugar, el punto de no retorno en el que ha habido un dao irreversible todava es en gran parte indeterminado; as, no tenemos un punto de corte preciso para separar causa y efecto. Finalmente, es posible que no haya una vafinalcomn nica por la que mueren las clulas. Por lo tanto, es difcil de finir el estadio ms all del cual la clula est irrecuperablemente condenada a la dest ruccin. Y cundo muere realmente la clula? Dos fenmenos caracterizan consisDURACIN DE L A AGRESIN tentemente la irreversibilidad. El primero es la incapacidad de reverti r la disfuncin mitocondrial (falta de fosforilacin oxidativa FIGURA 1-16 Cronologa de los cambios bioqumicos y moioM y produccin de ATP) incluso despus de la resolucin de la agregicos en la agresin celular. brana, causando potencialmente cambios en la permeabilidad y alteraciones electr ofisiolgicas17.
20 UNIDAD I f Patologa gengral microscopa ptica o examen macroscpico. Como podra esperarse, las manifestaciones mor folgicas de la necrosis tardan ms en desarrollarse que las del dao reversible. Por ejemplo, la tumefaccin celular es un cambio morfolgico reversible, y puede ocurrir en cuestin de minutos. Sin embargo, los cambios inconfundibles en un microscopio ptico de la muerte celular no ocurren en el miocardio hasta 4 a 12 horas tras la isquemia total, aunque sabemos que existe una lesin irreversible entre los 20 y 60 minutos. Lesin reversible Con el microscopio ptico pueden reconocerse dos patrones de lesin celular reversib le: tumefaccin celular y cambio graso. La tumefaccin celular aparece siempre que l as clulas son incapaces de mantener la homeostasia inica y de lquidos, y es el resu ltado de la prdida de funcin de las bombas inicas de la membrana plasmtica dependien tes de energa. El cambio graso ocurre en la lesin hipxica y en varias formas de les in txica o metablica. Se manifiesta por la aparicin de pequeas o grandes vacuolas lipd icas en el citoplasma y ocurre en la hipoxia y en diversas formas de lesin txica. Se encuentra principalmente en las clulas implicadas en y dependientes del metabo lismo graso, tales como el hepatocito y la clula miocrdica. Los mecanismos del cam bio graso se abordan con mayor detalle ms adelante en el captulo. Morfologa. La tumefaccin celular es la primera manifestacin de casi todas las forma s de agresin a las clulas. Es un cambio morfolgico difcil de apreciar con el microsc opio ptico; puede ser ms aparente desde el punto de vista de todo el rgano. Cuando afecta a muchas clulas en un rgano, produce algo de palidez, mayor turgencia y aum ento del peso de los rganos. Al examen microscpico pueden verse vacuolas pequeas cl aras dentro del citoplasma; representan segmentos distendidos y desprendidos del retculo endoplsmico. Este patrn de lesin no letal se denomina, a veces, cambio hidrp ico o degeneracin vacuolar. La hinchazn de las clulas es reversible. Los cambios es tructurales de la lesin celular reversible (Fig. 1-17) incluyen: 1. Alteraciones de la membrana plasmtica, tales como protrusiones, borrado y distorsin de las micr ovellosidades; creacin de figuras de mielina, y aflojamiento de las uniones inter celulares. 2. Cambios mitocondriales, que incluyen hinchazn, rarefaccin y la apari cin de pequeas densidades amorfas ricas en fosfolpidos. ~":.;;^ FIGURA 1-17 Cambios morfolgicos en lesin celular reversible e irreversible. A, mic rofotografa electrnica de una clula epitelial normal del tbulo renal proximal. Ntense las microvellosidades (mv) abundantes revistiendo la luz (L). N: ncleo; V: vacuo las apicales (que son estructuras normales en este tipo de clula). B, clula epitel ial del tbulo proximal mostrando cambios isqumicos reversibles. Se pierden las mic rovellosidades (mv) que se han incorporado al citoplasma apical; se han formado protrusiones que se expulsan a la luz (L). Las mitocondrias estn ligeramente dila tadas. (Comparar con A.) C, clula tubular proximal que muestra una lesin isqumica i rreversible. Ntense la mitocondrias marcadamente hinchadas que contienen densidad es amorfas, membranas celulares rotas y un ncleo picntico denso. (Cortesa del Dr. M .A. Venkatachalam, University of Texas, San Antonio, TX.)
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 21 3. Dilatacin del retculo endoplsmico, con desprendimiento y desagregacin de los poli somas. 4. Alteraciones nucleares, con desagregacin de los elementos granulares y fibrilares. Necrosis La necrosis se refiere a un espectro de cambios morfolgicos que sigue a la muerte celular en el tejido vivo como resultado, en gran medida, de la accin d egradante progresiva de las enzimas en la clula letalmente lesionada (las clulas f ijadas inmediatamente estn muertas pero no necrticas). Como se usa habitualmente, la necrosis es la correlacin macroscpica e histolgica de la muerte celular que ocur re en una situacin de lesin exgena irreversible. Las clulas necrticas son incapaces d e mantener la integridad de la membrana y, a menudo, sus contenidos se escapan. Esto puede ocasionar inflamacin en el tejido circundante. La apariencia morfolgica de la necrosis es el resultado de la desnaturalizacin de las protenas intracelula res y de la digestin enzimtica de la clula. Las enzimas se derivan de los lisosomas de las mismas clulas muertas, en cuyo caso la digestin enzimtica se denomina autlis is, o de los lisosomas de los leucocitos que han inmigrado durante las reaccione s inflamatorias. Estos procesos tardan horas en desarrollarse y, de esta manera, puede no haber cambios detectables en la clula si, por ejemplo, un infarto miocrd ico produce una muerte sbita. La nica evidencia manifiesta podra ser la oclusin de u na arteria coronaria. La evidencia histolgica ms precoz de la necrosis miocrdica no se hace manifiesta hasta 4 a 12 horas ms tarde, pero las enzimas especficas del c orazn y las protenas que se liberan del msculo necrtico pueden detectarse en sangre tan pronto como 2 horas tras la muerte de la clula miocrdica. mticas desnaturalizadas (Fig. 1-18). La clula necrtica puede tener una apariencia h omognea ms cristalina que la de las clulas normales, principalmente como resultado de la prdida de las partculas de glucgeno. Cuando las enzimas han digerido las orga nelas citoplasmticas, el citoplasma se hace vacuolado y aparece como comido por p olillas. Finalmente, puede ocurrir calcificacin de las clulas muertas. Al final, l as clulas muertas pueden sustituirse por grandes masas de fosfolpidos arremolinado s denominados figuras de mielina. Estos precipitados fosfolipdicos son entonces f agocitados por otras clulas o degradados ulteriormente a cidos grasos; la calcific acin de tales residuos de cidos grasos da lugar a la generacin de jabones de calcio . Por microscopia electrnica las clulas necrticas se caracterizan por discontinuida des manifiestas en la membrana plasmtica y de las organelas, marcada dilatacin de las mltocondrias con la apariencia de grandes densidades amorfas, figuras intrac itoplasmticas de mielina, residuos osmioflicos amorfos y agregados de material alg odonoso representando, probablemente, protena desnaturalizada (ver Fig. 1-17C). L os cambios nucleares aparecen en forma de uno de tres patrones, todos debidos a la fragmentacin inespecfica del DNA (ver Figs. 1 -8 y 1 -17C). La basofilia de la cromatina puede desvanecerse (carilisis), un cambio que posiblemente refleja la a ctividad DNasa. Un segundo patrn (que se ve tambin en la muerte celular apopttica) es la picnosis, caracterizada por encogimiento nuclear y aumento de la basofilia . Aqu el DNA aparentemente se condensa en una masa slida, encogida, basoflica. En e l tercer patrn, conocido como cariorrexis, los ncleos picnticos o parcialmente picnt icos sufren fragmentacin. Con el paso del tiempo (1 o 2 das), el ncleo de la clula n ecrtica desaparece totalmente. Una vez que las clulas necrticas han sufrido las pre coces alteraciones descritas, la masa de clulas necrticas puede tener varios patro nes morfolgicos. Aunque los trminos estn algo pasados de moda, se utilizan rutinari amente y los patlogos y los clnicos comprenden sus significados. Cuando la desnatu ralizacin es el patrn primario, se desarrolla una necrosis de coagulacin. En caso d e digestin enzimtica Morfologa. Las clulas necrticas muestran eosinofilia aumentada atribuible, en parte , a la prdida de la basofilia normal impartida por el RNA del citoplasma y, en pa rte, por el aumento de la unin de la eosina a las protenas intracitoplas-
FIGURA 1-18 Necrosis isqumica del miocardio. A, miocardio normal. B, miocardio co n necrosis de coagulacin (dos tercios superiores de la figura), mostrando fibras miocrdicas anucleadas fuertemente eosinoflicas. Los leucocitos en el intersticio f orman parte de la reaccin precoz al msculo necrtico. Comparar con A y con las fibra s normales en la parte inferior de la figura.
22 UNIDAD I % Patologa general dominante, el resultado es la necrosis por licuefaccin; en circunstancias especia les, puede haber necrosis caseosa y necrosis grasa. La necrosis por coagulacin im plica la conservacin del contorno bsico de la clula coagulada durante un perodo de a l menos algunos das (Fig. 1 -19/4). Los tejidos afectados muestran una textura fi rme. Presumiblemente, la lesin o la subsiguiente y creciente acidosis intracelula r desnaturaliza no solamente las protenas estructurales sino tambin las enzimas y, de esta manera, bloquea la protelisis de la clula. El infarto de miocardio es un excelente ejemplo en el cual las clulas acidfilas coaguladas anucleadas pueden per sistir durante semanas. Finalmente, las clulas miocrdicas necrticas se eliminan por fragmentacin y fagocitosis de los residuos celulares por los leucocitos limpiado res y por la accin de enzimas lisosomales proteolticas, tradas por los leucocitos q ue han imigrado. El proceso de necrosis por coagulacin, con conservacin de la arqu itectura general del tejido, es caracterstico de la muerte hipxica en todos los te jidos, excepto en el cerebro. La necrosis por licuefaccin es caracterstica de infe cciones bacterianas focales u, ocasionalmente, fngicas, ya que los microbios esti mulan la acumulacin de clulas inflamatorias (Fig. 1-198). Por razones no bien ente ndidas, la muerte hipxica de las clulas dentro del sistema nervioso central evoca, a menudo, la necrosis por licuefaccin. Cualquiera que sea la patognesis, la licue faccin digiere completamente las clulas muertas. El resultado final es la transfor macin del tejido en una masa viscosa lquida. Si el proceso se inici por una inflama cin aguda (Captulo 2), frecuentemente el material es amarillo cremoso por la prese ncia de leucocitos muertos y se denomina pus. Aunque la necrosis gangrenosa no e s un patrn distintivo de la muerte celular, el trmino todava se usa habitualmente e n la prctica clnica quirrgica. Habitualmente, se aplica a una extremidad, generalme nte la parte inferior de la pierna, que ha perdido su riego sanguneo y ha sufrido necrosis por coagulacin. Cuando la infeccin bacteriana se superpone, la necrosis por coagulacin se modifica por la accin licuefactiva de las bacterias y los leucocitos atrados (denominada gangrena hmeda) . La necrosis caseosa, una forma distintiva de necrosis por coagulacin, se encuen tra muy a menudo en los focos de infeccin tuberculosa (Captulo 8). El trmino caseos o se deriva de la apariencia macroscpica blanca, en aspecto de queso, del rea de n ecrosis (Fig. 1 -20). Al microscopio, en el foco necrtico aparecen residuos granu lares amorfos compuestos, aparentemente, de clulas fragmentadas, coaguladas y res iduos granulares amorfos rodeados por un reborde inflamatorio definido, conocido como reaccin granulomatosa (Captulo 2). A diferencia de la necrosis por coagulacin , la arquitectura tisular est completamente alterada. La necrosis grasa es un trmi no que est bien establecido en la jerga mdica pero no denota realmente un patrn esp ecfico de necrosis. Ms bien es descriptivo de reas focales de FIGURA 1-20 Pulmn tuberculoso con una gran rea de necrosis caseosa. Los residuos c aseosos son blanco-amarillos y con consistencia de queso. -- y \$*x&. B FIGURA 1-19 Necrosis de coagulacin y licuefaccin. A, infarto renal que exhibe una necrosis de coagulacin, con prdida de ncleos y formacin de grumos en el citoplasma p ero con conservacin de los contornos bsicos de la arquitectura glomerular y tubula r. 6, un foco de necrosis de licuefaccin en el rion producida por una infeccin fngic a. El foco est relleno con leucocitos y residuos celulares, creando un absceso re nal que altera la arquitectura normal.
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular 23 ruccin grasa, que ocurren tpicamente como resultado de liberacin de lipasas pancreti cas activadas en la sustancia I pncreas y en la cavidad peritoneal. Esto ocurre e n la encia abdominal calamitosa conocida como pancreatitis uda (Captulo 19). En e ste trastorno, las enzimas pancretiactivadas se escapan de las clulas y conductos acinares, enzimas activadas lican las membranas de las clulas adas, y las lipasas a ctivadas escinden los esteres de triglicridos contenidos dentro de las clulas gras as. Los cidos grasos liberados se combinan con el calcio para producir reas sos nc as, de aspecto de tiza, visibles macroscpicamente saponificacin de la grasa), que permiten al cirujano y al pat(sai logo identificar las lesiones (Fig. 1-21). En e l examen histolgi, la necrosis toma la forma de focos de clulas grasas necricas, co n contornos borrosos con depsitos basoflcos de calcio y rodeados de una reaccin infl amatoria. en algunas de estas situaciones, tiene caractersticas distintivas y, por lo tanto , se aborda ms adelante en el captulo.
LESIN ISQUMICA E HIPXICA ste es el tipo ms frecuente de lesin celular en medicina clnica y se ha estudiado ex tensamente en humanos, en animales de experimentacin y en sistemas de cultivo27"2 9. Han emergido escenarios razonables respecto a los mecanismos subyacentes a lo s cambios morfolgicos. La hipoxia se refiere a cualquier estado de disponibilidad reducida de oxgeno. Puede estar causada por cantidades disminuidas o por reduccin de la saturacin de la hemoglobina. La isquemia, por otra parte, sobreviene por u n riego sanguneo disminuido, habitualmente como consecuencia de una obstruccin mecn ica en el sistema arterial, pero, a veces, como resultado de una cada catastrfica en la presin sangunea o prdida de sangre. En contraste con la hipoxia, durante la c ual la produccin de energa glucoltica puede continuar, la isquemia compromete el su ministro de sustratos para la gluclisis. As pues, en los tejidos isqumicos, la gene racin de energa anaerbica se para despus de que se han consumido los sustratos gluco lticos, o la funcin glucoltica se inhibe por la acumulacin de metabolitos que, de ot ra manera, deberan haberse eliminado por el riego sanguneo. Por esta razn, la isque mia tiende a lesionar los tejidos ms rpido que la hipoxia. Tipos de lesin isqumica. La lesin isqumica es la expresin clnica ms frecuente de la lesin celular por la privac in de oxgeno. Los modelos ms tiles para estudiar la lesin isqumica implican la oclusin completa de una de las arterias terminales de un rgano (p. ej., una arteria coron aria) y el examen del tejido (p. ej., msculo cardaco) en las reas irrigadas por la arteria. Durante la isquemia ocurren cambios patolgicos complejos en los diversos sistemas celulares. Hasta cierto punto, con una duracin que vara con los diferent es tipos de clulas, la lesin es reparable, y las clulas afectadas pueden recuperars e si los sustratos metablicos y el oxgeno estn de nuevo disponibles con la restaura cin del riego sanguneo. Cuando la duracin de la isquemia se prolonga ms, la estructu ra celular contina deteriorndose, debido a la progresin incesante de los mecanismos lesivos en curso. Con el tiempo, la maquinaria energtica de la clula -la central oxidativa mitocondrial y la va glucoltica- se daa irreparablemente, y la restauracin del riego sanguneo (reperfusin) no puede rescatar la clula lesionada. Incluso cuan do la maquinaria energtica celular permanece intacta, un dao irreparable en el gen oma o en las membranas celulares dar lugar a un resultado letal, independientemen te de la reperfusin. Esta lesin irreversible se manifiesta, habitualmente, como ne crosis, pero la apoptosis tambin puede desempear un papel. En determinadas circuns tancias, cuando el riego sanguneo se restaura en las clulas que previamente han es tado en isquemia pero no han muerto, con frecuencia la lesin se exacerba paradjica mente y prosigue a un ritmo acelerado. Como consecuencia, los tejidos reperfundi dos pueden sufrir prdida de clulas adems de las clulas que estn irreversiblemente daad as al final de la isquemia. ste es un proceso clnicamente importante que contribuy e al dao tisular neto durante los infartos miocrdicos y celulares, como se aborda en los Captulos 12 y 28. Esta denominada lesin por isquemia-reperfusin (comentada ms
adelante) es particularmente significativa porque el tratamiento mdico apropiado puede disminuir la fraccin de clulas que, de otra manera, estaban destinadas a mo rir en el rea en riesgo. Mecanismos de lesin celular isqumica. La secuencia de suceso s tras la hipoxia se describi antes y se resume en la Figura 1 -2228'29. FIGURA 1-21 Focos de necrosis grasa con saponificacin en el mesenterio. Las reas d e depsitos blancos de aspecto de tiza representan formacin de jabn de calcio en los sitios de degradacin lipdica. Finalmente, en el paciente vivo, la mayora de las clulas necrticas y sus residuos d esaparecen con un proceso combinado de digestin enzimtica y fragmentacin, seguido d e fagocitosis de las partculas de residuos por los leucocitos. Si las clulas necrti cas y los residuos celulares no se destruyen y reabsorben rpidamente, tienden a a traer sales de calcio y otros minerales y a calcificarse. Este fenmeno, denominad o calcificacin distrfica, se comenta ms adelante en este captulo. Ejemplos de lesin y necrosis celulares Habiendo revisado brevemente las causas generales, mecanismos y morfologa de la l esin celular y de la muerte necrtica de la clula, ahora describimos algunas formas habituales de lesin celular, esto es, la lesin isqumica e hipxica, y algunos tipos d e lesin txica. Aunque la apoptosis contribuye a la muerte celular
24 UNIDAD I Patologa general va, probablemente, por liberacin de molculas proapoptticas de la mitocondria daada. Tras la muerte, los componentes celulares se degradan p rogresivamente, y existe un escape diseminado de las enzimas celulares al espaci o extracelular y, al revs, entrada de macromolculas extracelulares del espacio int ersticial a las clulas agonizantes. Finalmente, la clula muerta puede reemplazarse por grandes masas compuestas de fosfolpidos en forma de figuras de mielina. A co ntinuacin, stas son fagocitadas por otras clulas o degradadas posteriormente a cidos grasos. Puede haber calcificacin de estos residuos de cidos grasos con formacin de jabones de calcio. Como se mencion antes, el escape de enzimas intracelulares y otras protenas a travs de la membrana plasmtica, anormalmente permeable, y hacia el plasma proporciona parmetros clnicos importantes de muerte celular. Por ejemplo, los niveles sricos elevados de la creatincinasa MB y de troponina del msculo cardac o son unos valiosos indicadores clnicos de infarto de miocardio, un rea de muerte celular en el msculo cardaco (Captulo 12). LESIN DE ISQUEMIA REPERFUSION La restauracin del riego sanguneo a los tejidos isqumicos puede dar lugar a recuper acin de clulas si stas estn reversiblemente lesionadas, o no afecta al resultado cua ndo ha habido un dao celular irreversible. Sin embargo, dependiendo de la intensi dad y Si la isquemia persiste, se sigue de lesin irreversible y necrosis.duracin d e la agresin isqumica, un nmero variable de clulas La lesin irreversible se asocia, m orfolgicamente, con intensa tumepuede encaminarse hasta la muerte tras restaurars e el riego sanfaccin de las mitocondrias, dao extenso de las membranas plasguneo, m ediante necrosis as como apoptosis30. A menudo, los mticas e hinchazn de los lisoso mas (ver Fig. 1-17C). Se desatejidos afectados muestran infiltrados neutroflicos. Como se cit rrollan grandes densidades, amorfas floculantes, en la matriz anteri ormente, esta lesin de isquemia-reperfusin es un proceso mitocondrial. En el mioca rdio, hay indicios de lesin irreversible clnicamente importante en situaciones com o el infarto de mioque pueden verse tan pronto como de 30 a 40 minutos despus de cardio y el accidente vascular cerebral, y puede ser tratable. la isquemia. Ento nces ocurre un aflujo masivo de calcio hacia Cmo ocurre la lesin de reperfusin? La r espuesta probable dentro de las clulas, particularmente si la zona isqumica se per es que se establecen nuevos procesos lesivos durante la reperfusin, funde nuevame nte. La muerte acaece principalmente por necroproduciendo la muerte de las clulas que, de otra manera, podran sis, pero tambin contribuye la apoptosis; la va apoptti ca se actihaberse recuperado31,32. Se han propuesto varios mecanismos: LESIN REVE RSIBLE LESIN IRREVERSIBLE (muerte celular) Lesin de la membrana Prdida de fosfolpidos Alteraciones citoesquelt lcas Radicales libres Degradacin de lpidos Otros t Escape enzimtico (CK, LDH) De forma breve, segn disminuye la presin de oxgeno dentro de la clula, hay una prdida de fosforilacin oxidativa y generacin disminuida de ATP. La deplecin del ATP da lu gar a la insuficiencia de la bomba de sodio, con prdida de potasio, aflujo de sod io y agua, y tumefaccin celular. Hay una prdida progresiva de glucgeno y una dismin ucin de la sntesis proteica. Puede haber varias consecuencias funcionales graves e n este estadio. Por ejemplo, el msculo cardaco deja de contraerse a los 60 segundo s de oclusin coronaria. Sin embargo, ntese que la prdida de contractilidad no signi fica muerte celular. Si se contina la hipoxia, el empeoramiento de la deplecin del ATP produce un deterioro morfolgico ulterior. El citoesqueleto se dispersa, dand o lugar a la prdida de caractersticas estructurales tales como las microvellosidad es y la formacin de burbujas en la superficie celular (ver Fig. 1-17). Pueden verse figuras de mielina, derivadas de las membranas plasmticas y de las organelas dentr o del citoplasma o extracelularmente. Se piensa que son el resultado de la disoc iacin de lipoprotenas, poniendo al descubierto grupos fosfatdicos, favoreciendo la captacin e intercalacin de agua entre las capas laminares de las membranas. En est e momento, las mitocondrias habitualmente estn hinchadas debido a la prdida del co ntrol del volumen por estas organelas; el retculo endoplsmico permanece dilatado, y toda la clula est marcadamente hinchada, con concentraciones aumentadas de agua, sodio y cloro, y una concentracin disminuida de potasio. Si se restablece el oxge
no, todas estas alteraciones son reversibles. \ f Entrada de Ca 2+ Mitocondria t Fosforilacin oxidativa Bomba . deNa + t Entrada de Ca 2+ H 2 0, yNa + Salida de K+ Tumefaccin celular Prdida de microvellosidades Protrusiones Hinchazn del RE Figuras de mielina Agrupamiento de cromatina nuclear tATP Otros efectos t Gluclisis i Glucgeno Liberacin intracelular y activacin de enzimas lisosomales tBasofilia(RNP) Cambios nucleares Digestin proteica \ Desprendimiento de ribosomas t Sntesis de protena Depsito lipdico FIGURA 1-22 Secuencia postulada de acontecimientos en la lesin celular isqumica re versible e irreversible. Ntese que, aunque la reduccin de la fosforilacin oxidativa y de los niveles de ATP tiene un papel central, la isquemia puede producir dao d irecto en la membrana. RE: retculo endoplsmico; CK: creatincinasa; LDH: lactato de shidrogenasa; RNP: ribonucleoprotena.
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 25 El nuevo dao puede iniciarse durante la reoxigenacin por la La mayora de los dems ag entes qumicos no son biolgicageneracin aumentada de radicales libres de oxgeno por e l parn- mente activos, sino que deben convertirse en metabolitos txicos o reactivo s que, entonces, actan sobre las clulas diana. quima y las clulas endoteliales y de los leucocitos infiltrantes31,33. Esta modificacin se consigue, habitualmente, p or las oxidasas Los aniones superxido pueden producirse en el tejido reirrigaP450 de funcin mixta, en el retculo endoplsmico liso del do como resultado de una reduc cin incompleta y vicariante de hgado y de otros rganos37,38. Aunque estos metabolit os pueoxgeno por las mitocondrias daadas o por la accin de oxidaden causar dao en la membrana y lesin celular mediante una sas derivadas de los leucocitos, clulas end oteliales o clulas unin covalente directa a la protena y lpidos de la membrana, pare nquimatosas32. Los mecanismos celulares de defensa antiel mecanismo ms importante con diferencia de la lesin de la oxidante tambin pueden estar afectados por la is quemia, favomembrana implica la formacin de radicales libres reactivos reciendo l a acumulacin de radicales. Los limpiadores de radicay la peroxidacin lipdica subsig uiente. les libres pueden ser teraputicamente beneficiosos. Las especies de oxgeno reactivo pueden, ulteriormente, Los diversos mecanismos de lesin qumica quedan bi en ilusfavorecer la transicin de la permeabilidad mitocondrial, referitrados por el tetracloruro de carbono y el paracetamol. El tetrada anteriormente, que, cuan do ocurre, impide la recuperacin cloruro de carbono (CC14) se utiliz ampliamente e n tiempos en la energtica mitocondrial y la recuperacin celular de ATP, industria de limpieza en seco39. El efecto txico del CC14 se debe a y conduce a la muerte c elular25. La lesin isqumica se asocia con inflamacin como resulta- su conversin medi ante el P450 al radical libre txico altamente reactivo CC13 (CCL, + e ~ CC13 + CL) (Fig. 1-23). Los radicales do de la produccin de citocinas y expresin aumentada d e molculas de adhesin por el parnquima hipxico y las clulas endoteliales31'33. Estas molculas recluan leucocitos poliCCI 4 morfonucleares circulantes para tejido reper fundido; la infla1 REL macin subsiguiente produce lesin adicional (Captulo 2). La i mportancia del aflujo de neutrfilos en la lesin reperfundida CCI| se ha demostrado con estudios experimentales que han utilizaI cido graso polienoico microsomal do tratamientos antiinflamatorios, tales como anticuerpos Radicales lipideos antici tocinas o antimolculas de adhesin, para reducir la amplitud de la lesin30,32. l+02 Datos recientes sugieren que la activacin de la va del complePEROXIDACIN DE LQUIDOS mento puede contribuir a la lesin de isquemia-reperfusin34. El Diseminacin autocata ltica a lo largo sistema del complemento est implicado en la defensa del husde la m embrana microsomal ped y es un mecanismo importante de lesin inmunitaria (Captulo 6). Por razones desconocidas, algunos anticuerpos IgM tienDao de la Liberacin de p roductos den a depositarse en los tejidos isqumicos y, cuando se restablece membr ana del RER de peroxidacin lipdica el riego sanguneo, las protenas del complemento s e unen a los anticuerpos, se activan y producen lesin celular e inflamacin. Los ra tones defectivos (knockouf), que carecen de varias protenas Dao en la membrana del complemento, son resistentes a este tipo de lesin35. plasmtica T LESIN QUMICA Los mecanismos por los que agentes qumicos, ciertos frmacos y toxinas producen les in se describen con mayor detalle en el Captulo 9, en la exposicin de la enfermedad ambiental. Aqu describimos dos formas de lesin inducida qumicamente como ejemplos de la secuencia de acontecimientos que conducen a la muerte celular. Los agentes qumicos inducen lesin celular por uno de dos mecanismos generales36: Algunos agen tes qumicos pueden actuar directamente combinndose con algn componente molecular crt ico o con una organela celular. Por ejemplo, en el envenenamiento por cloruro de mercurio, el mercurio se une a los grupos sulfidrilo de la membrana celular y d e otras protenas, produciendo un aumento de la permeabilidad de la membrana e inh ibicin del transporte dependiente de ATPasa. En tales situaciones, el mayor dao se produce, habitualmente, en las clulas que usan, absorben, excretan o concentran los agentes qumicos -en el caso del cloruro de mercurio, las clulas del tracto gas
trointestinal y del rion (Captulo 9)-. El cianuro envenena la citocromo oxidasa mi tocondrial y bloquea la fosforilacin oxidativa. Muchos agentes quimioterpicos anti neoplsicos y frmacos antibiticos tambin inducen un dao celular mediante efectos citotx icos directos. Desprendimiento del polisoma 1 i 1 1 \ * Sntesis de apoprotena 1 Permeabilidad de Na+, H 2 0, Ca 2+ \ Tumefaccin celular Entrada masiva de Ca 2+ Hgado graso Inactivacin de mitocondrias, enzimas celulares y desnaturalizacin de protenas FIGURA 1-23 Secuenci a de acontecimientos que dan lugar a la degeneracin grasa y a la necrosis celular en la toxicidad por el tetracloruro de carbono (CCI4). REL : retculo endoplsmico liso; RER: retculo endoplsm co rugoso.
26 UNIDAD I f Patologa general libres producidos localmente producen autooxidacin de los citotoxicidad se correla ciona con la peroxidacin lipdica y puede dos grasos polienoicos presentes dentro d e los fosfolpidos de reducirse con la administracin de antioxidantes, sugiriendo q ue, en la toxicidad principal del frmaco41, el dao oxidativo puede membrana. All se inicia la descomposicin oxidativa del lpido y ser ms importante que la unin covalen te. se forman perxidos orgnicos tras reaccionar con oxgeno (peroxidacin lipdica). Est a reaccin es autocatalticaya que los nuevos radicales se forman a partir de los pr opios radicales perxidos. La Apoptosis rpida rotura de la estructura y de la funcin del retculo endoLa apoptosis es una va de muerte celular inducida por un prograpls mico se debe a la descomposicin del lpido. Por lo tanto, no sorprende el hecho de que la lesin de la clula heptica inducida ma intracelular estrechamente regulado en el cual las clulas destinadas a morir activan enzimas que degradan el propio DNA de por CCl4 es grave y, adems, de comienzo extremadamente rpido. la clula y las pr otenas nucleares y citoplasmticas. La membrana En menos de 30 minutos hay una dism inucin en la sntesis proplasmtica de las clulas permanece intacta, pero su estructur a teicoheptica; a las 2 horas, hay una tumefaccin del retculo est alterada, de tal m anera que la clula apopttica se transforma endoplsmico liso y disociacin de los ribo somas del retcuen un blanco vido para la fagocitosis. La clula muerta se elimilo en doplsmico rugoso. La exportacin lipdica desde los hepana rpidamente, antes de que su contenido se escape, y por lo tocitos se reduce debido a su incapacidad de sint etizar apoprotetanto la muerte celular por esta va no suscita una reaccin inflana p ara unirse con los triglicridos y, por lo tanto, facilitar la matoria en el husped . As pues, la apoptosis es fundamentalsecrecin lipoproteica. El resultado es el hga do graso de la intoximente diferente de la necrosis, que se caracteriza por prdid a de la cacin por CCI4 (Fig. 1-24). (El hgado graso se comenta despus integridad de membrana, digestin enzimtica de las clulas en este captulo.) La lesin mitocondrial o curre a continuacin, y y, frecuentemente, una reaccin del husped (ver Fig. 1 -9 y T ase sigue de progresiva tumefaccin de las clulas debida a una perbla 1-2). Sin emb argo, la apoptosis y la necrosis coexisten a veces, meabilidad aumentada de la m embrana plasmtica. Se piensa que y pueden compartir algunas caractersticas y mecan ismos. el dao de la membrana plasmtica est producido por aldehidos grasos relativam ente estables, que se producen por la peroxidacin lipdica en el retculo endoplsmico liso pero que son capaCAUSAS DE APOPTOSIS ces de actuar a distancia. Esto es seg uido por el aflujo masivo de calcio y muerte celular (ver Fig. 1-23). La apoptos is se reconoci inicialmente en 1972 por su morfologa distintiva, y se denomin segn l a designacin griega para cada o decaimiento42. Ocurre normalmente en muchas situacio nes, y sirve para eliminar clulas indeseadas o potencialmente dainas y las clulas q ue ya no son tiles. Asimismo, es un acontecimiento patolgico cuando las clulas estn daadas sin remisin, especialmente cuando el dao afecta a su DNA; en estas situacion es, la clula irremediablemente daada se elimina. La apoptosis es responsable de nu merosos acontecimientosfisiolgicos,adaptativos y patolgicos, enumerados a continua cin. Apoptosis en situaciones fisiolgicas La muerte por apoptosis es un fenmeno nor mal que sirve para eliminar clulas que ya no se necesitan, como por ejemplo duran te el desarrollo, y para mantener un nmero fijo de diversas poblaciones celulares en los tejidos. Es importante en las siguientes situaciones fisiolgicas: Destruc cin programada de las clulas durante la embriognesis, incluyendo la implantacin, org anognesis, involucin durante el desarrollo y metamorfosis. El trmino muerte celular programada se acu originalmente para denotar la muerte de tipos celulares especficos en tiempos determinados durante el desarrollo43. La apoptosis es un trmino genric o para este modo de muerte celular, independientemente del contexto, pero a menu do se utiliza indistintamente con muerte celular programada. Involucin hormonodepen diente en el adulto, como la rotura celular endometrial durante el ciclo menstru al, la atresia folicular ovrica en la menopausia, la regresin de la mama lactante tras el destete, y la atrofia prosttica tras la castracin. Eliminacin celular en la
s poblaciones celulares proliferativas, como el epitelio de las criptas intestin ales, afinde mantener un nmero constante. Muerte de clulas del husped que han cumpl ido su propsito, como los neutrfilos en una respuesta inflamatoria aguda, y los li nfocitos alfinalde una respuesta inmunitaria. En estas situacioEl paracetamol, un frmaco analgsico frecuentemente utilizado, se desintoxica en el hgado por la sulfatacin y glucuronizacin, y pequeas cantidades son convertidas, por la oxidacin catalizada por el citocromo P450, a un metabolito electroflico altame nte txico40. A su vez, este metabolito se desintoxica por interaccin con el GSH. C uando se ingieren grandes dosis del frmaco, el GSH se agota y, entonces, los meta bolitos txicos se acumulan en la clula, destruyen las macromolculas nucleoflicas, y se unen covalentemente protenas y cidos nucleicos. La disminucin en la concentracin de GSH, junto con la unin covalente de los metabolitos txicos, aumenta la toxicida d del frmaco, dando lugar a una necrosis masiva de la clula heptica, habitualmente de 3 a 5 das despus de la ingestin de la dosis txica. Esta hepa-
CAPITULO '' Adaptaciones celulares, lesin celular y muerte celular 27 nes, las clulas sufren apoptosis porque carecen de las seales de supervivencia nec esarias, tales como los factores de crecimiento. Eliminacin de linfocitos autorre activos potencialmente dainos, bien antes o bien despus de que han completado su m aduracin (Captulo 6). Muerte celular inducida por linfocitos T citotxicos, un mecan ismo de defensa contra los virus y tumores que sirve para eliminar las clulas inf ectadas por virus y las neoplsicas. El mismo mecanismo es responsable del rechazo celular en los trasplantes (Captulo 6). Apoptosis en situaciones patolgicas La mu erte por apoptosis tambin es responsable de la prdida de clulas en diversos estados patolgicos: Muerte celular producida por una variedad de estmulos lesiFIGURA 1-25 Caractersticas ultraestructurales de la apoptosis. vos. Por ejemplo, la radiacin y los frmacos antineoplsicos Algunos fragmentos nucleares muestran semilunas perifr icas de citotxicos daan el DNA y, si los mecanismos de reparacin cromatina compacta da, mientras que otros son uniformemente no dan abasto con la lesin, las clulas se destruyen a s mismas densos. (De Kerr JFR, Harmon BV: Definition and incidence o f apoptosis: a historical perspective. En Tomei LD, Cope FO [eds]: por apoptosis . En estas situaciones, la eliminacin de la clula Apoptosis: The Molecular Basis o f Cell Death. Cold Spring Harbor, puede ser una alternativa mejor que el riesgo de mutaciones y NY, Cold Spring Harbor Laboratory Press, 1991, pp 5-29.) trasloc aciones en el DNA daado, que puede dar lugar a transformacin maligna. Estos estmulo s lesivos, adems del calor y de la hipoxia, pueden inducir apoptosis si la agresin es leve, pero las dosis elevadas del mismo estmulo dan lugar a la muerte celular por necrosis. El estrs del retculo endoplsmi Formacin de protrusiones citoplasmticas y cuerpos co (RE), que est inducido por acumulacin de protenas desplegadas, tambin d esencadena la muerte apopttica de la apoptticos. La clula apopttica muestra, primera mente, clula (descrito ms adelante en el captulo). formacin extensa de protrusiones en la superficie, a continuacin sufre fragmentacin en cuerpos apoptticos li Lesin cel ular en ciertas enfermedades vricas, tales como hepatitis vrica, en las cuales la prdida de las clulas infectadas gados a la membrana, compuestos de citoplasma y or gase debe, en su mayor parte, a la muerte apopttica. nelas empaquetadas estrecham ente, con o sin fragmentos Atrofia patolgica en los rganos parenquimatosos tras ob snucleares. truccin ductal, como ocurre en el pncreas, glndula partida Fagocitosis d e las clulas apoptticas o cuerpos celulay rion. res, habitualmente por los macrfagos . Los cuerpos Muerte celular en tumores, ms frecuentemente durante la apoptticos s e degradan rpidamente dentro de los lisosoregresin pero tambin en los tumores en cr ecimiento activo. mas, y las clulas sanas adyacentes migran o proliferan para Segn se mencion antes, incluso en situaciones en que la reemplazar el espacio ocupado por la clula apopttica muerte celular se debe principalmente a necrosis, la va de ahora suprimida. la apoptosis puede contribuir. Por ejemplo, estmulos lesivos que producen aumento de la permeabilidad en las mitoconSe piensa que las membranas plasmticas permanecen drias desencadenan la apoptosis. Antes de que se aborden lo s mecanismos de la apoptosis, describimos las caractersticas morfolgicas y bioqumic as de este proceso. intactas durante la apoptosis, hasta los ltimos estadios en los que se hacen perm eables a solutos normalmente retenidos. Esta descripcin clsica es precisa respecto a la apoptosis durante condiciones fisiolgicas tales como la embriognesis y la el iminacin de clulas inmunitarias. Sin embargo, tras estmulos agresivos, no son infre cuentes formas de muerte celular con caractersticas de necrosis as como de apoptos is44. Sin embargo, en tales condiciones, la intensidad, ms que la especificidad, del estmulo determina la forma en la cual se expresa la muerte. Si las caractersti cas necrticas son predominantes, ocurre el dao precoz de la membrana plasmtica, y s e ve tumefaccin celular ms que encogimiento. En el examen histolgico de los tejidos teidos con hematoxilina y eosina, la apoptosis afecta a clulas aisladas o a pequea s agrupaciones celulares. La clula apopttica aparece como una masa redondeada u ov al de citoplasma intensamente eosinoflico, con fragmentos densos de cromatina nuc
lear (Fig. 1-26). Dado que el encogimiento celular y la forMorfologa. Las siguientes caractersticas morfolgicas, algunas observables mejor con el microscopio electrnico, caracterizan a las clulas que sufren apoptosis (Fig. 1 -25). Encogimiento celular. La clula tiene menor tamao; el citoplasma es denso; la s organelas, aunque relativamente normales, estn empaquetadas ms estrechamente. Co ndensacin de la cromatina. sta es la apariencia ms caracterstica de la apoptosis. La cromatina se agrega perifricamente, debajo de la membrana nuclear, en masas dens as de varias configuraciones y tamaos. El propio ncleo puede romperse, produciendo dos o ms fragmentos.
28 UNIDAD I * Patologa general FIGURA 1-26 A, apoptosis de clulas epidrmicas en una reaccin mediada inmunolgicament e. Las clulas apoptticas son visibles en la epidermis con citoplasma intensamente eosinoflico y ncleos pequeos, densos. Tincin H&E. (Cortesa del Dr. Scott Granter, Bri gham and Womens Hospital, Boston, MA.) 6, gran aumento de una clula apopttica en e l hgado con una lesin celular mediada inmunolgicamente. (Cortesa del Dr. Dhanpat Jai n, Yale University, New Haven, CT.) macin de cuerpos apoptticos son rpidos, y los fragmentos son fagocitados rpidamente, puede haber una considerable apoptosis en los tejidos antes de que se haga apar ente en los cortes histolgicos. Adems, la apoptosis -a diferencia de la necrosisno desencadena inflamacin, hacindose ms difcil de detectar histolgicamente. CARACTERSTICAS BIOQUMICAS DE LA APOPTOSIS Las clulas apoptticas habitualmente muestr an un conjunto distintivo de modificaciones bioqumicas que subyacen a los cambios estructurales descritos anteriormente. Algunas de estas caractersticas tambin pue den verse en las clulas necrticas, pero otras alteraciones son ms especficas. Escisin de protena. Una caracterstica especfica de la apoptosis es la hidrlisis proteica im plicada en la activacin de varios miembros de una familia de proteasascistenicas, denominadas caspasas43. Muchas caspasas estn presentes en las clulas nrmales como p roenzimas inactivas, y necesitan activarse para inducir apoptosis. Las caspasas activas escinden muchas protenas celulares vitales, tales como lminas, y as rompen el armazn nuclear y el citoesqueleto; adems, las caspasas activan las DNasas, que degradan el DNA nuclear. Estos cambios subyacen en las alteraciones estructurale s nucleares y citoplasmticas que ocurren en las clulas apoptticas. Fragmentacin del DNA. Las clulas apoptticas exhiben una fragmentacin caracterstica del DNA en grandes fragmentos de 50 a 300 kilobases46. Subsiguientemente, hay una escisin internucl eosomal del DNA a oligonucleosomas, en mltiplos de 180 a 200 pares de bases, por las endonucleasas dependientes de Ca2+ y Mg2+. Los fragmentos pueden visualizars e mediante electroforesis en gel de agarosa como escaleras de DNA (Fig. 1 -27). La actividad FIGURA 1-27 Electroforesis en gel de agarosa del DNA extrado de clulas en cultivo. Tincin con bromuro de etidio; fotografiado con iluminacin ultravioleta. Lnea A, cu ltivo control. Lnea B, cultivo de clulas expuestas al calor mostrando apoptosis ex terna; ntese el patrn en escalera de los fragmentos de DNA, que representa mltiplos de oligonucleosomas. Lnea C, cultivo que muestra una necrosis masiva; ntese la ex tensin difusa del DNA. El patrn en escalera est producido por la escisin enzimtica de l DNA nuclear en fragmentos de tamao del nucleosoma, habitualmente mltiplos de 180 -200 pares de bases, fsfos patrones son caractersticos de la apoptosis y de la ne crosis, respectivamente, pero no especficos de ellas. (De Kerr JFR, Harmon BV: De finition and incidence of apoptosis: a historical perspective. En Tomei LD, Cope FO [eds]: Apoptosis: The Molecular Basis of Cell Death. Cold Spring Harbor. NY, Cold Spring Harbor Laboratory Press, 1991, p 13.)
CAPITULO Adaptaciones celulares, lesin celular y muerte celular 29 de la endonucleasa tambin es la base de la deteccin de la muerte celular mediante tcnicas citoqumicas que reconocen roturas de la doble cadena de DNA47. Sin embargo , la escisin internucleosomal del DNA no es especfica de la apoptosis. Se piensa q ue el patrn difuso de la (smeared) fragmentacin del DNA en un gel es indicativo de n ecrosis, pero esto puede ser un fenmeno autoltico tardo, y las tpicas escaleras de D NA pueden verse, adems, en las clulas necrticas47. Reconocimiento fagoctico. Las clul as apoptticas expresan fosfatidilserina en las capas externas de sus membranas pl asmticas, en las que los fosfolpidos se han dado la vuelta hacia fuera desde las cap as internas. (A causa de estos cambios, las clulas apoptticas pueden identificarse por la fijacin de colorantes especficos tales como Annexin V.) En algunos tipos d e apoptosis, la trombospondina, una glucoprotena de adhesin, tambin se expresa en l as superficies de los cuerpos apoptticos, y otras protenas segregadas por los fago citos pueden unirse a las clulas apoptticas y opsonizarlas para su fagocitosis48. Estas alteraciones permiten el reconocimiento precoz de la muerte celular por lo s macrfagos, dando lugar a fagocitosis sin liberacin de componentes celulares proi nflamatorios49. De esta manera, la respuesta apopttica dispone de las clulas con afectacin mnima del tejido circundante. MECANISM OS DE LA APOPTOSIS La apoptosis se induce por una cascada de acontecimientos mol eculares que se inicia de distintas maneras y culmina con la activacin de caspasa s (Fig. 1-28)45. Puesto que se piensa que un exceso o un defecto de apoptosis su byace en muchas enfermedades, como las enfermedades degenerativas y el cncer, exi ste un gran inters en dilucidar los mecanismos de esta forma de muerte celular. S e han hecho tremendos progresos en nuestro conocimiento sobre la apoptosis. Uno de los hechos notables que emergen es que los mecanismos bsicos de apoptosis estn conservados en todos los metazoos50. De hecho, algunos de los principales avance s derivan de las observaciones hechas en el nematodo Caenorhabditis elegans, cuy o desarrollo sigue el procedimiento de un patrn altamente reproducible de crecimi ento celular seguido de muerte celular. Los estudios de gusanos mutantes han per mitido la identificacin de genes especficos (denominados genes ced, que significa muerte celular anormal) que inician o inhiben la Va intrnseca (mitocondrial) Va extrnseca (muerte iniciada en el receptor) . . , ... Protrusion citoplasmatica Cuerpo apopttico FIGURA 1-28 Mecanismos de apoptosis. Los marcados (1) son algunos de los mayores inductores de apoptosis. Incluyen los ligandos especficos de muerte (factor de n ecrosis tumoral [TNF] y ligandos Fas), la retirada de factores de crecimiento u hormonas, y agentes lesivos (p. ej., radiacin). Algunos estmulos (tales como clulas citotxicas) activan directamente las caspasas de ejecucin {derecha). Otros actan p or medio de protenas adaptadoras y caspasas iniciadoras, o por acontecimientos mi tocondriales que implican al citocromo c. (2) El control y la regulacin estn influ idos por miembros de la familia de protenas Bcl-2, que pueden inhibir o favorecer la muerte de la clula. (3) Las caspasas ejecutoras activan proteasas y endonucle asas latentes del citoplasma que degradan las protenas del ncleo y citoesqueleto. Esto da lugar a una cascada en la degradacin intracelular, incluyendo la fragment acin de la cromatina nuclear y rotura del citoesqueleto. (4) El resultado final e
s la formacin de cuerpos apoptticos que contienen organelas ntracelulares y otros c omponentes citoslicos; estos cuerpos tambin expresan nuevos ligandos para unirse y ser captados por clulas fagocticas.
30 UNIDAD I # Patologa general apoptosis y para los que existen homlogos definidos en los mamferos. El proceso de apoptosis puede dividirse en una fase de iniciacin, durante la cual las caspasas se hacen catalticamente activas, y la fase de ejecucin, durante la cual esas enzi mas actan produciendo muerte celular. La iniciacin de la apoptosis ocurre principa lmente por seales procedentes de dos vas distintas pero convergentes: la va extrnsec a o iniciada en el receptor, y la intrnseca o mitocondrial. Ambas vas convergen en la activacin de caspasas. Describimos estas dos vas por separado porque implican interacciones moleculares bastante distintas, pero es importante recordar que es tn interconectadas en numerosos pasos. Va extrnseca (muerte iniciada en el receptor ). Esta va se inicia por la implicacin del receptor de muerte de la superficie cel ular en una diversidad de clulas51. Los receptores de muerte son miembros de la f amilia del receptor del factor de necrosis tumoral que contiene un dominio citop lsmico implicado en las interacciones protena-protena que se denomina el dominio de muerte, dado su papel esencial en el suministro de seales apoptticas. (Algunos mi embros de la familia del receptor de TNF no contienen dominios citoplsmicos de mu erte; su papel en el desencadenamiento de la apoptosis est mucho menos establecid o.) Los receptores de muerte mejor conocidos son el receptor de TNF de tipo 1 (T NFR1) y una protena relacionada denominada Fas (CD95), pero se han descrito otros diversos. El mecanismo de apoptosis inducido por estos receptores de muerte se ilustra bien con Fas (Fig. 1-29). Cuando Fas se une cruzadamente por medio de su ligando, el ligando Fas unido a la membrana (FasL), tres o ms molculas de Fas se juntan y sus dominios de muerte citoplasmticos forman un sitio de unin para una pr otena adaptadora que contiene tambin un dominio de muerte denominado FADD (Fas-ass ociated death domain). El FADD que se une a los receptores de muerte se une, a s u vez, a una forma inactiva de la caspasa-8 (y, en humanos, caspasa-10), otra ve z a travs de un Caspasas ejecutoras \ APOPTOSIS FIGURA 1-29 Va extrnseca (muerte iniciada en el receptor) de la apoptosi s, ilustrada por acontecimientos que siguen a la unin de Fas (ver texto). dominio de muerte. As pues, mltiples molculas pro-caspasa-8 se aproximan, y se esci nden unas a otras para generar caspasa-8 activa. Entonces, la enzima desencadena una cascada de activacin de caspasa escindiendo, y as activando, otras pro-caspas as, y las enzimas activas median en la fase de ejecucin de la apoptosis (comentad a despus). Esta va de apoptosis puede inhibirse por una protena denominada FLIP, qu e se une a la pro-caspasa-8, pero no puede escindir y activar la enzima porque l e falta la actividad enzimtica52. Algunos virus y clulas normales producen FLIP y utilizan este inhibidor para proteger a las clulas infectadas y normales de la ap optosis mediada por Fas. El esfingolpido ceramida se ha implicado como un interme diario entre los receptores de muerte y la activacin de caspasa, pero su papel en esta va no est claro y sigue siendo controvertido53. Va intrnseca (mitocondrial). E sta va de apoptosis es el resultado de una permeabilidad mitocondrial aumentada y liberacin de molculas pro-apoptticas al citoplasma sin intervencin de los receptore s de muerte34'55. Los factores de crecimiento y otras seales de supervivencia est imulan la produccin de miembros antiapoptticos de la familia de protenas Bcl-256. E sta familia se denomina a partir del Bcl-2, que se identific como un oncogn en lin fomas de clulas B y es homlogo a la protena Ced-9 de C. elegans. Existen ms de 20 pr otenas en esta familia, todas las cuales funcionan regulando la apoptosis; las do s antiapoptticas principales son Bcl-2 y Bcl-x. Estas protenas antiapoptticas resid en normalmente en las membranas mitocondriales y en el citoplasma. Cuando las clu las no reciben de seales de supervivencia o estn sujetas a estrs, la membrana mitoc
ondrial pierde Bcl-2 y/o Bcl-x que se sustituyen por miembros pro-apoptticos de l a familia, tales como Bak, Bax y Bim. Cuando disminuyen los niveles de Bcl-2/Bcl -x, la permeabilidad de la membrana mitocondrial aumenta, y se escapan varias pr otenas que pueden activar la cascada de la caspasa (Fig. 1-30). Una de esas proten as es el citocromo c, bien conocido por su papel en la respiracin mitocondrial. E n el citosol, el citocromo c se une a una protena denominada Apaf-1 {apoptosis ac tivating factor-1, homlogo a Ced-4 de C. elegans), y el complejo activa a la casp asa-957. (Bcl-2 y Bcl-x pueden, asimismo, inhibir directamente la activacin de Ap af-1, y su prdida por las clulas puede permitir la activacin de Apaf-1.) Otras prot enas mitocondriales, tales como el factor inductor de apoptosis (AIF), penetran e n el citoplasma donde se unen, y neutralizan, a diversos inhibidores de la apopt osis, cuya funcin normal es bloquear la activacin de caspasas'8. El resultado neto es la iniciacin de una cascada de caspasa. As pues, la esencia de esta va intrnseca es un equilibrio entre molculas pro-apoptticas y protectoras que regulan la perme abilidad mitocondrial y la liberacin de inductores de muerte que estn normalmente secuestrados dentro de la mitocondria. Existe una considerable evidencia de que la va intrnseca de la apoptosis puede desencadenarse sin que intervengan las mitoc ondrias59. La apoptosis puede iniciarse mediante la activacin de caspasa previa a la mitocondria, y el aumento subsiguiente en la permeabilidad mitocondrial y la liberacin de molculas proapoptticas amplifican la seal de muerte46. Sin embargo, es as vas de apoptosis que implican la iniciacin independiente de las mitocondrias no estn bien definidas. Hemos descrito las vas extrnsecas e intrnsecas de iniciacin de la apoptosis como distintas, pero puede haber superposiciones entre ellas. Por e jemplo, en los hepatocitos, la seal de Fas activa un miembro pro-apopttico de la f amilia Bel,,denominado Bid, que a su vez activa la va mitocondrial. No se sabe si esas interacciones cooperativas entre las vas apoptticas estn activas en la mayora de los dems tipos celulares.
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 31 CITOSOL Membrana interna Otras protenas pro-apoptticas (p. ej., AIF) Citocromo c Se une a y neutraliza Inhibidores de apoptosis (IAP) Apaf-1 Activacin de caspasa MATRIZ MITOCONDRIAL FIGURA 1-30 Va intrnseca (mitocondrial) de la apoptosis. Los agonistas de la muert e producen cambios en la membrana mitocondrial interna, dando lugar a la transic in de la permeabilidad mitocondrial (TPM) y a la liberacin de citocromo c y otras protenas pro-apoptticas en el citosol, que activan las caspasas (ver texto). Membr ana externa Caspasas ejecutoras #APOPTOSIS Fase de ejecucin. La fasefinalde la apoptosis est mediada por una cascada proteolti ca, hacia la cual convergen los diversos mecanismos de iniciacin. Las proteasas q ue median la fase de ejecucin se conservan en gran medida en las distintas especi es y pertenecen a la familia de la caspasa, segn se ha mencionado previamente. So n homologas en mamferos del gen ced-3 de C. elegansu. El trmino caspasa se basa en dos propiedades de esta familia de enzimas: la c se refiere a una proteasa cisteni ca (es decir, una enzima con cistena en su sitio activo), y aspasa se refiere a la capacidad singular de estas enzimas para escindir a continuacin los residuos de ci do asprtico60. La familia caspasa, que incluye actualmente ms de diez miembros, pu ede dividirse funcionalmente en dos grupos bsicos -iniciador y ejecutordependiend o del orden en que se activan durante la apoptosis60. Las caspasas iniciadoras, como hemos visto, incluyen caspasa-8 y caspasa-9. Varias caspasas, entre ellas c aspasa-3 y caspasa-6, sirven como ejecutoras. Como muchas proteasas, las caspasa s existen como proenzimas inactivas, o zimgenos, y deben sufrir una escisin activa dora para que se inicie la apoptosis. Las caspasas tienen sus propios sitios de escisin que pueden hidrolizarse no solamente por otras caspasas sino tambin autoca talticamente. Despus de que una caspasa iniciadora es escindida para producir su f orma activa, el programa de muerte enzimtica se pone en movimiento por la activac in rpida y secuencial de otras caspasas. Las caspasas ejecutoras actan sobre muchos componentes celulares. Escinden el citoesqueleto y las protenas de la matriz nuc lear y, de esta manera, rompen el primero y dan lugar a la fragmentacin del ncleo6 0. En el ncleo, las dianas de la activacin de caspasas incluyen protenas implicadas en la transcripcin, replicacin y reparacin del DNA. En particular, la activacin de la caspasa-3 convierte a una DNasa citoplasmtica en una forma activa escindiendo un inhibidor de la enzima; esta DNasa induce la caracterstica escisin internucleos omal del DNA, descrita anteriormente. Eliminacin de clulas muertas. En los primero s estadios de la apoptosis, las clulas moribundas segregan factores solubles que reclutan los fagocitos61. Esto facilita la eliminacin rpida de clulas apoptticas ant es de que sufran necrosis secundaria y liberacin de sus contenidos celulares (que puede dar lugar a inflamacin). Como ya se ha referido, las clulas apoptticas y sus fragmentos tienen molcul as marcadoras en su superficie que facilitan el reconocimiento precoz por las clu las adyacentes o fagocitos para la captacin y eliminacin fagoctica. Se ha demostrad o que numerosos receptores macrofgicos estn implicados en la unin e ingestin de las clulas apoptticas. Adems, los macrfagos tambin pueden segregar sustancias que se unen especficamente a las clulas apoptticas, pero no a las vivas, y las opsonizan para su fagocitosis. En contraste con los marcadores en clulas apoptticas, parece que l as clulas viables evitan su propia ingestin por los macrfagos mediante la expresin d e ciertas molculas de superficie (tales como CD31). Este proceso de fagocitosis d e clulas apoptticas es tan eficiente que las clulas muertas desaparecen sin dejar r astro y la inflamacin est virtualmente ausente. EJEMPLOS DE APOPTOSIS Las seales qu
e inducen apoptosis incluyen la falta de factor u hormona de crecimiento, la imp licacin especfica de los receptores de muerte y agentes lesivos concretos. Aunque el ejemplo clsico de apoptosis ha sido la muerte programada de clulas durante la e mbriognesis, todava no sabemos lo que desencadena la apoptosis en esta situacin. Si n embargo, se conocen muchos otros ejemplos de apoptosis bien definidos. Apoptos is tras privacin de factor de crecimiento. Las clulas sensibles a hormonas privada s de la correspondiente hormona, los linfocitos que no estn estimulados por antgen os y citocinas y las neuronas privadas de factor de crecimiento nervioso mueren por apoptosis62. En todas estas situaciones, la apoptosis est desencadenada por l a va intrnseca (mitocondrial) y es atribuible a un exceso de miembros pro-apopttico s de la familia Bel en relacin con miembros antiapoptticos. Apoptosis mediada por dao del DNA. La exposicin de clulas a radiacin o a agentes quimioterpicos induce la a poptosis por un mecanismo que se inicia con el dao al DNA (estrs genotxico) y que i mplica al gen supresor tumoral p536>. p53 se acumula cuando el DNA est daado y det iene el ciclo celular (en la fase Gj) para dar tiempo a su reparacin (Captulo 7). Sin
32 UNIDAD I % Patologa general embargo, si falla el proceso de reparacin del DNA, p53 desencadena la apoptosis. Cuando p53 est mutado o ausente (como ocurre en ciertos cnceres), es incapaz de in ducir apoptosis y favorece la supervivencia celular. As pues, parece que p53 sirv e como un conmutador crtico vida o muerte en caso de estrs genotxico. El mecanismo po r el cual p53 desencadena la maquinaria efectora mortal distal -las caspasas- es complejo, pero su bien caracterizada funcin de activacin de la transcripcin parece estar implicada. Entre las protenas cuya produccin est estimulada por p53 hay vari os miembros pro-apoptticos de la familia Bel, especialmente Bax y Bak, as como Apa f-1, mencionados antes. Estas protenas activan las caspasas y producen apoptosis. Trastornos asociados con apoptosis defectuosa y aumento de la supervivencia celu lar. Aqu, una tasa inadecuadamente baja de apoptosis puede prolongar la supervive ncia o reducir el recambio de clulas anormales. Estas clulas acumuladas pueden dar lugar a: (1) cnceres, especialmente tumores con mutaciones de p53, o tumores dep endientes de hormonas, tales como cnceres de mama, prstata u ovario (Captulo 7), y (2) trastornos autoinmunitarios, que pueden surgir si los linfocitos autorreacti vos no se eliminan tras encontrarse con autoantgenos (Captulo 6). Trastornos asoci ados con aumento de apoptosis y muerte celular excesiva. Estas enfermedades se c aracterizan por una marcada prdida de clulas normales o protectoras e incluyen: (1 ) enfermedades neurodegenerativas, manifestadas por prdida de Apoptosis inducida por la familia de receptores del factor de grupos especficos de neuronas, tales como atrofias musculares necrosis tumoral. Como se expuso antes, el receptor de superficie espinales (Capt ulo 27); (2) lesin isqumica, como en el infarcelular Fas (CD95) induce apoptosis c uando se le une su ligando to de miocardio (Captulo 12) y el accidente vascular c erebral (FasL o CD95L), que es producido por clulas del sistema inmu(Captulo 28), y (3) muerte de clulas infectadas por virus, en nitario. Este sistema es importan te en la eliminacin de linfocitos muchas infecciones vricas (Captulo 8). que recono cen autoantgenos, y las mutaciones en Fas o FasL dan lugar a enfermedades autoinm unitarias en humanos y ratones Respuestas subcelulares a la lesin (Captulo 6)64. L a citocina TNF es un importante de la reaccin inflamatoria Hasta este punto en el captulo la atencin se ha enfocado sobre la (Captulo 2), pero tambin es capaz de ind ucir apoptosis. clula como una unidad. Sin embargo, ciertos trastornos se aso(El nombre factor de necrosis tumoral surgi no porque la citocian con alteraciones dist intivas en las organelas celulares o en el cina destruya directamente las clulas tumorales, sino porque citoesqueleto. Algunas de estas alteraciones coexisten co n las desinduce trombosis de los vasos sanguneos tumorales, dando lugar critas en la lesin letal aguda; otras representan formas ms crnia la muerte isqumica del tumo r.) La unin de TNF a TNFR1 cas de lesin celular; an otras son respuestas de adaptac in que conduce a la asociacin del receptor con la protena adaptadora sirven para ma ntener la homeostasia. Aqu tratamos solamente TRADD (protena que contiene TNF rece ptor-associated death algunas de estas reacciones ms frecuentes o ms interesantes. domairi). A su vez, TRADD se une a FADD y da lugar a la apoptosis a travs de la activacin de caspasa, lo mismo que las interacCATABOLISMO LISOSOMAL ciones Fas-Fa sL52. Sin embargo, las principales funciones del TNF no estn mediadas por la indu ccin de la apoptosis sino por la Los lisosomas primarios son organelas intracelul ares unidas a la activacin del importante factor de transcripcin factor KB nuclear membrana que contienen una diversidad de enzimas hidrolticas, (NF-KB). Las seales mediadas por TNF realizan esto estimulanincluyendo fosfatasa acida, glucuronida sa, sulfatasa, ribonucleasa do la degradacin del inhibidor de NF-KB (IKB) 65 . El sistema y colagenasa. Estas enzimas se sintetizan en el retculo endoplsregulador de la transcripcin NF-KB/IKB es importante para la mico rugoso y despus se empaque tan dentro de vesculas, en el supervivencia celular y, como veremos en el Captulo
2, para aparato de Golgi. Los lisosomas primarios se unen a las vacuolas numeros as respuestas inflamatorias. Dado que el TNF puede ligadas a la membrana, vacuol as que contienen material para inducir la muerte celular y promover la supervive ncia de la cludigerir, formando lisosomas secundarios o fagolisosomas. Los lisola , qu determina este ying y yang de su accin? La respuesta no somas estn implicados e n la fragmentacin del material fagoest clara, pero probablemente dependa de qu prot ena adaptacitado de dos posibles formas: heterofagia y autofagia (Fig. 1-31). dor a se una al receptor de TNF tras fijar la citocina. TRADD y FADD favorecen la ap optosis, y otras protenas adaptadoras Heterofagia. La heterofagia es el proceso d e la digestin lisodenominadas TRAF (factores asociados al receptor TNF) favoresom al de materiales ingeridos del ambiente extracelular. Los cen la activacin de NFKB y la supervivencia. materiales extracelulares son captados por las clulas medi ante el proceso general de endocitosis. La captacin de partculas Apoptosis mediada por linfocito T citotxico. Los linfocitos T de material se conoce como fagocitos is; la captacin de macrocitotxicos (LTCs) reconocen los antgenos ajenos presentados molculas solubles menores se denomina pinocitosis. Los matesobre la superficie d e clulas infectadas del husped (Captulo 6). riales extracelulares son introducidos por endocitosis en En el reconocimiento, los LTCs segregan perforina, una molcula vacuolas (endosomas o fagosomas) que,finalmente,se funden formadora de poros tr ansmembrana, que permite la entrada de la con los lisosomas para formar fagoliso somas, donde el material serina proteasa del granulo del LCT, denominada granzim a B. La ingerido se digiere. La heterofagia es ms frecuente en los fagogranzima B tiene la capacidad de escindir protenas en los resicitos profesionales, tales como neutrfilos y macrfagos, duos aspartato y tambin de activar diversas caspasas celul ares66. De esta manera, el LTC destruye las clulas diana cortocircuitando aunque tambin puede ocurrir en otros tipos celulares. Ejemlas seales precedentes e induci endo directamente la fase efectora deplos de heterofagocitosis incluyen la capta cin y digestin de bacterias por neutrfilos y la eliminacin de clulas apopttila apoptos is. Los LTCs tambin expresan FasL en su superficie y cas por macrfagos. destruyen las clulas diana unindose a los receptores Fas, como se describi antes. Autofagia. La autofagia se refiere a la digestin lisosomal de los propios componentes de la clula. En este proceso, las orgaSe ha postulado la apoptosis disregulada (demasiad o o demanelas intracelulares y porciones del citosol son primeramente siado poco) para explicar componentes de un amplio rango de secuestradas del citoplasma en una vacuola autofgica formada enfermedades67. En esencia, dos grupos de trastorno s pueden ser por regiones del retculo endoplsmico rugoso libres de riboel resultad o de esta disregulacin:
CAPITULO 1 # Adaptaciones celulares, lesin celular y muerte celular HETEROFAGIA Lisosoma primario AUTOFAGIA Lisosoma primario 33 carbono inhaladas de la atmsfera o pigmentos inoculados en los tatuajes, pueden p ersistir en los fagolisosomas de los macrfagos durante dcadas. Los lisosomas tambin son sitios de depsito en los que las clulas secuestran sustancias anormales que n o pueden metabolizarse completamente. Los trastornos hereditarios de almacenamie nto lisosomal causados por deficiencias de enzimas que degradan diversas macromo lculas, dan lugar a la acumulacin de cantidades anormales de esos compuestos en lo s lisosomas de las clulas por todo el cuerpo, particularmente las neuronas, produ ciendo anomalas graves (Captulo 5). Ciertos frmacos pueden alterar la funcin lisosom al y producir enfermedades lisosmicas adquiridas o inducidas por frmacos {iatrognic as). Los frmacos de este grupo incluyen cloroquina, un agente antimalrico que elev a el pH interno del lisosoma, inactivando as sus enzimas. Inhibiendo las enzimas lisosomales, la cloroquina reduce el dao tisular en las reacciones inflamatorias, que estn mediadas, en parte, por enzimas liberadas por los leucocitos; esta accin es la base del uso del frmaco en enfermedades autoinmunitarias como la artritis reumatoide. Sin embargo, la misma inhibicin de las enzimas puede dar lugar a la a cumulacin anormal de glucgeno y fosfolpidos en los lisosomas, produciendo una miopa ta txica. INDUCCIN (HIPERTROFIA) DEL RETCULO ENDOPLSMICO LISO El RE liso est implicado en el metabolismo de diversos productos qumicos, y las clulas expuestas a estos p roductos muestran hipertrofia del RE como una respuesta de adaptacin que puede te ner consecuencias funcionales importantes. El uso prolongado de barbitricos da lu gar a un estado de tolerancia, con una disminucin en los efectos del frmaco y la n ecesidad de utilizar dosis crecientes. Se dice que los pacientes se han adaptado a la medicacin. Esta adaptacin se debe a un volumen aumentado (hipertrofia) del RE liso de los hepatocitos, que metaboliza el frmaco (Fig. 1-32). Los barbitricos se modifican en el hgado mediante una desmetilacin oxidativa, que implica al sistema oxidasa P450 de funcin mixta que se encuentra en el RE liso. El papel de estas mo dificaciones enzimticas es aumentar la solubilidad de una somas. La vacuola se funde con lisosomas o elementos de Golgi para formar un aut ofagolisosoma6*'69. La autofagia es un fenmeno frecuente implicado en la eliminac in de organelas daadas durante la lesin celular y en la remodelacin celular de la di ferenciacin, y es particularmente pronunciada en clulas que sufren atrofia inducid a por privacin de nutrientes o involucin hormonal. Las enzimas en los lisosomas so n capaces de degradar la mayora de las protenas e hidratos de carbono, pero alguno s lpidos permanecen sin digerir. Los lisosomas con residuos no digeridos pueden p ersistir dentro de la clula como cuerpos residuales o pueden expulsarse. Los gran ulos del pigmento lipofuscina representan material no digerido derivado de la pe roxidacin lipdica intracelular. Ciertos pigmentos no digeribles, como partculas de FIGURA 1-32 Microfotografa electrnica del hgado de rata tratada con fenobarbital qu e muestra un marcado aumento en la cantidad de retculo endoplsmico. (De Jones AL, Fawcett DW: Hypertrophy of the agranular endoplasmic reticulum in hmster liver in duced by Phenobarbital. J Histochem Cytochem 14:215, 1966. Cortesa del Dr. Fawcet t.)
34 UNIDAD I Patologa general variedad de compuestos (p. ej., alcohol, esteroides, eicosanoides y carcingenos, as como insecticidas y otros contaminantes ambientales) y as facilitar su secrecin3 7. Aunque a menudo se piensa que esto es una forma de desintoxicacin, muchos compue stos se tornan ms lesivos por la modificacin de P450. Adems, los productos formados por este metabolismo oxidativo incluyen formas de oxgeno reactivo, que pueden pr oducir lesin de la clula. Con el uso prolongado, los barbitricos (y muchos otros ag entes) estimulan la sntesis de ms enzimas, as como ms RE liso. De esta manera, la clu la tiene mejor capacidad para modificar los agentes qumicos y adaptarse a su ambi ente alterado. Las clulas adaptadas a un medicamento tienen capacidad aumentada p ara metabolizar otros compuestos manejados por el sistema. As, si los pacientes q ue toman fenobarbital para la epilepsia aumentan su ingestin de alcohol pueden te ner niveles subteraputicos de la medicacin anticonvulsivante por la induccin del RE liso en respuesta al alcohol. ALTERACIONES MITOCONDRIALES Hemos visto que la di sfuncin mitocondrial desempea un importante papel en la lesin celular y la apoptosi s. Adems, en algunas afecciones patolgicas, ocurren diversas alteraciones en el nme ro, tamao y forma de las mitocondrias. Por ejemplo, en la hipertrofia y atrofia c elulares, existen un aumento y una disminucin, respectivamente, en el nmero de mit ocondrias en las clulas. Las mitocondrias pueden asumir formas extremadamente gra ndes y anormales (megamitocondria), como puede verse en el hgado de la hepatopata alcohlica y en ciertas deficiencias nutricionales (Fig. 1-33). Actualmente, se sa be que las anomalas de las mitocondrias son la base de muchas enfermedades gentica s70 (Captulo 5). En ciertas enfermedades hereditarias del msculo esqueltico, las mi opatas mitocondriales, los defectos en el metabolismo mitocondrial se asocian con una mayor cantidad de mitocondrias que, a menudo, son anormalmente grandes, tie nen crestas anormales y contienen cristaloides (Captulo 27). Adems, ciertos tumore s benignos encontrados en las glndulas salivales, tiroides, paratiroides y rones co nsisten en clulas (a veces denominadas oncocitos) con abundantes mitocondrias agran dadas, dando a la clula una apariencia eosinoflica distintiva. ANOMALAS CITOESQUELETICAS Las anomalas del citoesqueleto subyacen en varios estado s patolgicos. El citoesqueleto consta de microtbulos (de 20 a 25 nm de dimetro), fi lamentos delgados de actina (de 6 a 8 nm), filamentos gruesos de miosina (15 nm) , y varias clases de filamentos intermedios (10 nm). Tambin existen varias otras formas de protenas contrctiles no polimerizadas y no filamentosas. Las anomalas cit oesquelticas pueden ponerse de manifiesto por: (1) defectos en la funcin celular, como la locomocin celular y los movimientos intracelulares de organelas, y (2) en algunos casos, por acumulaciones intracelulares de material fibrilar. Se citan solamente unos pocos ejemplos: Filamentos delgados. Los filamentos delgados estn compuestos de actina, miosina y sus protenas reguladoras asociadas71. Los filamen tos delgados funcionantes son esenciales en varios estadios del movimiento leuco citario o en la capacidad de estas clulas de llevar a cabo adecuadamente la fagoc itosis. Algunos frmacos y toxinas tienen por diana losfilamentosde actina y, de e sta manera, afectan a estos procesos. Por ejemplo, la citocalasina B evita la po limerizacin de los filamentos de actina y la faloidina, una toxina del hongo Aman ita phaloides, tambin se une a losfilamentosde actina. Microtbulos. Defectos en la organizacin de los microtbulos pueden inhibir la motilidad del esperma, producien do esterilidad masculina, y pueden inmovilizar los cilios del epitelio respirato rio, produciendo interferencia con la capacidad de este epitelio para eliminar b acterias inhaladas, dando lugar a bronquiectasias (sndrome de Kartagener, o sndrom e del cilio inmvil; Captulo 15). Los microtbulos, como los microfilamentos, son ese nciales para la migracin y fagocitosis de los leucocitos. Los agentes qumicos, tal es como la colchicina, se unen a la tubulina y evitan la alineacin de los microtbu los. Este agente se utiliza en las crisis agudas de gota para evitar la migracin de leucocitos y la fagocitosis en respuesta al depsito de cristales de urato. Los microtbulos son un componente esencial del huso mittico, que se requiere para la divisin celular. Los frmacos que se unen a los microtbulos (p. ej., alcaloides de l
a vinca) pueden ser antiproliferativos y, por lo tanto, actan como agentes antitu morales. Filamentos intermedios. Estos componentes proporcionan un andamiaje int racelular flexible que organiza el citoplasma y resiste las fuerzas aplicadas a la clula72. Los filamentos intermedios se dividen en cinco clases, incluyendo fil amentos de queratina (caractersticos de clulas epiteliales), neurofilamentos (neur onas), filamentos de desmina (clulas musculares), filamentos de vimentina (clulas del tejido conectivo) y filamentos guales (astrocitos). La acumulacin de filament os de queratina y neurofilamentos se asocia con ciertos tipos de lesin celular. P or ejemplo, el cuerpo de Mallory, o hialina alcohlica, es una inclusin intracitoplas mtica eosinoflica en los hepatocitos caracterstica de la hepatopata alcohlica73, aunq ue puede estar presente en otros procesos. Tales inclusiones estn compuestas, pre dominantemente, de filamentos intermedios de queratina (Fig. 1-34). En el sistem a nervioso, los neurofilamentos estn presentes en el axn, donde proporcionan un ap oyo estructural. El ovillo neurofibrilar que se encuentra en el cerebro en la en fermedad de Alzheimer contiene protenas asociadas a microtbulos y neurofilamentos, que son un reflejo de una rotura del citoesqueleto neuronal (Captulo 28). Las mu taciones en los genes de filamentos intermedios pueden producir mltiples trastorn os humanos, incluyendo miopatas, enfermedades neurolgicas y dermopatas. FIGURA 1-33 Mitocondria agrandada con morfologa anormal del hgado de un paciente c on cirrosis alcohlica. Ntense, asimismo, las formaciones cristalinas en la mitocon dria.
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular 35 FIGURA 1-34 A, hgado por abuso de alcohol (alcoholismo crnico). Las inclusiones hi alinas en la clula del parnquima heptico en el centro aparecen como una red eosinofl ica dispuesta alrededor del ncleo {flecha). B, microfotografa electrnica de hialina alcohlica. El material est compuesto de filamentos intermedios (prequeratina) y u na matriz amorfa. La mayora del nfasis sobre la funcin del citoesqueleto se ha 2. Una sustancia endgen a normal o anormal se acumula por de ejercido en su papel mecnico, manteniendo la arquitectura celutos genticos o adquiridos en el metabolismo, empaquetamie lar, en la adhesin celular y la locomocin. Recientemente, se ha transporte o secrecin de estas sustancias. Un ejemplo es el gru reconocido que las protenas del citoesque leto estn conectadas con de trastornos producido por defectos genticos de enzimas muchos receptores celulares, tales como receptores linfocitarios especficas impli cadas en el metabolismo de lpidos e hidratos para antgenos, y son participantes ac tivos en la transduccin de de carbono dando lugar al depsito intracelular de estas susseal por estos receptores (Captulo 3). Por lo tanto, los defectos tancias, en gran parte en los lisosomas. Las as llamadas enferen las uniones entre los recept ores y las protenas del citoesquelemedades de almacenamiento se comentan en el Ca ptulo 5. to pueden afectar a muchas respuestas celulares. El sndrome de Otro es la deficiencia en alfa,-antitripsina, en la cual la sustituWiskott-Aldrich es una enfermedad hereditaria caracterizada por cin de un nico aminocido en la enzima da l ugar a defectos eccema, anomalas plaquetarias e inmunodeficiencia. La protena en e l plegamiento de la protena y acumulo de la enzima en el que est mutada en esta en fermedad est implicada en la unin de retculo endoplsmico del hgado en forma de inclus iones gloreceptores antignicos linfocitarios (y quizs otros receptores) al bulares eosinoflicas (ver ms adelante y Captulo 18). citoesqueleto, y los defectos en la p rotena interfieren con diversas 3. Una sustancia exgena anormal se deposita y se a cumula por respuestas celulares (Captulo 6)74. que la clula no tiene ni la maquina ria enzimtica para degradar la sustancia ni la capacidad de transportarla a otros sitios. Los acmulos de partculas de carbn y productos quAcmulos intracelulares micos no metabolizables, como las partculas de slice, son Una de las manifestaciones de los trastornos metablicos en las ejemplos de este tipo de alteracin. clulas es la acumulacin intracelular de cantidades anormales de diversas sustancias. Las susta ncias acumuladas se dividen en tres Sea cual sea la naturaleza u origen del acum ulo intracelular, categoras: (1) un constituyente celular normal acumulado en imp lica el almacenamiento de algn producto por las clulas indiv exceso, como agua, lpi dos, protenas e hidratos de carbono; (2) duales. Si la sobrecarga se debe a un tr astorno sistmico y puede una sustancia anormal, ya sea exgena, como mineral o prod uctos controlarse, la acumulacin es reversible. En las enfermedades de agentes in fecciosos, o endgena, como un producto de sntesis genticas de almacenamiento la acu mulacin es progresiva, y las o metabolismo anormal, y (3) un pigmento. Estas sust ancias pue- clulas pueden llegar a estar tan sobrecargadas que llega a produden a cumularse transitoria o permanentemente, y pueden ser cirse una lesin secundaria, dando lugar, en algunos casos, a la inocuas para las clulas pero, en ocasiones, son altamente txicas. muerte del tejido y del paciente. La sustancia puede locali zarse en el citoplasma (frecuentemente dentro de los fagolisosomas) o en el ncleo . En algunos casos, la LPIDOS clula puede estar produciendo la sustancia anormal, y en otros puede ser meramente un almacn de productos de procesos paTodas las cla ses principales de lpidos pueden acumularse en las tolgicos que ocurren en otro si tio del cuerpo. clulas: triglicridos, colesterol/steres de colesterol y fosfolpidos. Muchos procesos dan lugar a acmulos intracelulares anormaLos fosfolpidos son comp onentes de lasfigurasde mielina que se les, pero la mayora de ellos son atribuibl es a tres tipos de anomaencuentran en las clulas necrticas. Adems, complejos anorma las (Fig. 1-35): les de lpidos e hidratos de carbono se acumulan en las enfermedad es lisosmicas de almacenamiento (Captulo 5). Aqu nos centramos en las acumulaciones de triglicridos y colesterol. 1. Una sustancia endgena normal se produce a un rit
mo normal o aumentado pero el del metabolismo es inadecuado para eliminarla. Un ejemplo de este tipo de proceso es el cambio Esteatosis (cambio graso) graso en el hgado por la acumulacin intracelular de triglicridos (ver ms adelante). Otro es l a aparicin de gotitas de Los trminos esteatosis y cambio graso describen los acmulo s protena reabsorbida en los tbulos renales por un escape anormales de triglicridos dentro de las clulas parenquimatosas. aumentado de protena en los glomrulos. A men udo, el cambio graso se ve en el hgado porque es el rgano
36 UNIDAD I Patologa general Clula normal SQ O Hgado graso o Mutacin proteica 0 Plegamiento proteico, transporte - X Diferentes mecanismos-son responsables de la acumulacin de triglicridos en el hgado . Los cidos grasos libres del tejido adiposo o del alimento ingerido se transport an normalmente a los hepatocitos. En el hgado se esterifican a triglicridos, se co nvierten en colesterol o fosfolpidos, o se oxidan a cuerpos cetnicos. Algunos cidos grasos tambin se sintetizan a partir del acetato. La liberacin de triglicridos por los hepatocitos requiere la asociacin con apoprotenas para formar lipoprotenas, qu e entonces pueden viajar por la circulacin (Captulo 4). El exceso de acumulacin de triglicridos en el hgado puede ser el resultado de defec en cualquiera de los acon tecimientos de la secuencia, desde la entr da de cido graso a la salida de lipopr otena (Fig. 1-36A). Varios de estos defectos estn inducidos por el alcohol, una he patotoxina que altera las funciones mitocondriales y microsomales. El CC14 y la malnutricin proteica actan disminuyendo la sntesis de apoprotenas. La anoxia inhibe la oxidacin de cidos grasos. La inaAcidos grasos libres CAPTACIN cidos grasos Falta de enzima O w _l i Acetato Oxidacin a cuerpos cetnicos, CO2 a-glicerofosfato V ^ ^ - Fosfolpidos o Sustrato ^~ productos complejo L j ^ # '"solubles v Enzima > 'Sustrato complejo ^L^. /^^ > Esteres de colesterol i m Triglicridos Apoprotena O
/ Enfermedad de almacenamiento lisosmico: acumulacin de materiales endgenos o Ingestin de materiales no digeribles Lipoprotenas 1 SECRECIN Acumulacin de lpidos Acumulacin de materiales exgenos FIGURA 1-35 Mecanismos de acumulaciones ntracelula res: (1) metabolismo anormal, como en la degeneracin grasa del hgado; (2) mutacion es que producen alteraciones en el plegamiento de las protenas y su transporte, c omo en el dficit de cxrantitripsina; (3) deficiencia de enzimas crticas que evitan la degradacin de sustratos que se acumulan en los Msosomas, como en las enfermed ades lisosmicas de almacenamiento, y (4) incapacidad de degradar partculas fagocit adas, como en la hemosiderosis y en la acumulacin del pigmento del carbn. FIGURA 1 -36 Hgado graso. A, diagrama esquemtico de posibles mecanismos que dan lugar a la acumulacin de triglicridos ms importante implicado en el metabolismo de las grasas, pero en el hgado graso. Los defectos en alguno de los pasos de captacin, cataboli smo o secrecin pueden dar lugar a la acumulacin tambin ocurre en el corazn, msculo y rion. Las causas de lipdica. B, detalle a gran aumento del cambio graso del hgado. En esteatosis incluyen toxinas, malnutricin proteica, diabetes mellibien conserva do est compritus, obesidad y anoxia. En los pases industrializados, la causa msla m ayora de las clulas, el ncleodel citoplasma alrededor de la mido contra un anillo d esplazado frecuente, con diferencia, degeneracin grasa significativa del hgavacuol a grasa. (6, cortesa del Dr. James Crawford, Department of do (hgado graso) es el abuso de alcohol (Captulo 18)75. Pathology, Yale University School of Medicine, N ew Haven, CT.)
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular 37 nicin aumenta la movilizacin de cidos grasos desde los almacenes perifricos. El sign ificado de la degeneracin grasa depende de la causa y la intensidad de la acumula cin. Cuando es leve, puede no tener efectos sobre la funcin celular. El cambio gra so ms grave puede empeorar la funcin celular, tpicamente cuando algunos procesos in tracelulares vitales tambin estn obstaculizados (p. ej., en la intoxicacin por CC14 ). En una forma grave de lesin, la degeneracin grasa puede ser un precursor de la muerte celular. En aos recientes se han reconocido la esteatohepatitis no alcohlic a y la hepatopata grasa no alcohlica como entidades bastante comunes que pueden da r lugar a cirrosis e incluso cncer hepatocelular (Captulo 18). ms oscuro, rojo-marrn, no afectado (efecto atigrado). El otro patrn de hipoxia est p roducido por una hipoxia ms profunda o por alguna forma de miocarditis (p. ej., d ifteria) y muestra miocitos ms uniformemente afectados. Colesterol y esteres de colesterol El metabolismo celular del colesterol (abordado con detalle en el Captulo 5) est e strechamente regulado, de tal manera que la mayora de las clulas utilizan el coles terol para la sntesis de membranas celulares sin acumulacin intracelular de colest erol o de sus esteres. Sin embargo, las acumulaciones, manifestadas histolgicamen te como vacuolas intracelulares, se ven en varios procesos patolgicos: Ateroscler osis. En las placas aterosclerticas, las clulas musculares lisas y los macrfagos de ntro de la capa ntima de la aorta y de las grandes arterias estn rellenas de vacuo las lipdicas, la mayora de las cuales estn formadas por colesterol y sus esteres. T ales clulas tienen una apariencia espumosa (clulas espumosas), y su acumulo en la n tima produce los ateromas amarillos cargados de colesterol, caractersticos de est e serio trastorno. Algunas de estas clulas cargadas de grasa se rompen, liberando lpidos en el espacio extracelular. Los mecanismos de la acumulacin de colesterol en ambos tipos celulares en la aterosclerosis se comentan en detalle en el Captul o 11. Los esteres de colesterol extracelular pueden cristalizar en forma de aguj as largas, produciendo hendiduras muy caractersticas en las secciones tisulares. Xantomas. La acumulacin intracelular de colesterol dentro de los macrfagos tambin e s caracterstica de los estados hiperlipidmicos adquiridos y hereditarios. Se encue ntran agrupaciones de clulas espumosas en el tejido conectivo subepitelial de la piel y tendones, produciendo masas tumorales conocidas como xantomas. Inflamacin y necrosis. Los macrfagos espumosos se encuentran frecuentemente en sitios de les in celular e inflamacin, debido a la fagocitosis de colesterol de las membranas de las clulas lesionadas, incluyendo clulas parenquimatosas, leucocitos y eritrocito s. En los focos inflamatorios tambin se encuentran fosfolpidos y figuras de mielin a. Cuando son abundantes, los macrfagos cargados de colesterol proporcionan una c oloracin amarillenta en estos focos inflamatorios. Colesterolosis. Este trmino se refiere al acumulo focal de macrfagos cargados de colesterol en la lmina propia de la vescula biliar (Fig. 1-37). Se desconoce el mecanismo de acumulacin. Enfermeda d de Niemann-Pick tipo C. En esta enfermedad lisosmica de almacenamiento est mutad a una enzima implicada en el trfico de colesterol y, por ello, el colesterol se a cumula en mltiples rganos (Captulo 5). PROTENAS Morfologa. La degeneracin grasa se ve ms a menudo en el hgado y en el corazn. En todo s los rganos, el cambio graso aparece como vacuolas claras dentro de las clulas de l parnquima. Las acumulaciones intracelulares de agua o polisacridos (p. ej., glucg eno) tambin pueden producir vacuolas claras, y es necesario recurrir a tcnicas esp eciales para distinguir estos tres tipos de vacuolas claras. La identificacin de lpidos requiere evitar los disolventes de grasas comnmente utilizados para la incl usin en parafina para las tinciones rutinarias de hematoxilina y eosina. Para ide ntificar la grasa.es necesario preparar los cortes congelados de tejido a partir
de tejidos frescos o fijados con formol acuoso. Las secciones pueden teirse a co ntinuacin con Sudn IV u Oil Red-0, y ambos darn un color naranja-rojo a los lpidos c ontenidos. Finalmente, se emplea la reaccin de cido perydico-Schiff (PAS) para iden tificar el glucgeno, aunque no es especfica en absoluto. Cuando no pueden demostra rse ni grasa ni polisacridos en una vacuola clara, se presume que contiene agua o lquido con un contenido bajo en protenas. Hgado. En el hgado la degeneracin grasa le ve puede no afectar a la apariencia macroscpica. Con la acumulacin progresiva, el r gano se agranda y se hace cada vez ms amarillo hasta que, en circunstancias extre mas, el hgado puede llegar a pesar de 3 a 6 kg y transformarse en un rgano amarill o, blando y grasiento. La degeneracin grasa empieza con el desarrollo de inclusio nes diminutas, ligadas a la membrana (liposomas), pegadas estrechamente al retcul o endoplsmico. La degeneracin grasa se ve primeramente con el microscopio ptico com o pequeas vacuolas en el citoplasma alrededor del ncleo. Al progresar el proceso, las vacuolas se funden, creando espacios claros que desplazan al ncleo hasta la p eriferia de la clula (Fig. 1 -366). Ocasionalmente, las clulas contiguas se rompen , y los glbulos de grasa incluidos se funden produciendo los denominados quistes de grasa. Corazn. Los lpidos se encuentran en el msculo cardaco en forma de pequeas g otitas, que ocurren con dos patrones. En uno, la hipoxia prolongada moderada, ta l como la producida por una anemia profunda, produce depsitos intracelulares de g rasa, que crean una apariencia macroscpica de bandas de miocardio amarillento alt ernando con bandas de miocardio Por lo general, la acumulacin intracelular de protenas se manifiesta como gotitas eosinoflicas redondeadas, vacuolas o agregados en el citoplasma. Al microscopio e lectrnico pueden tener una apariencia amorfa, fibrilar o cristalina. En algunos p rocesos, tales como ciertas formas de amiloidosis, las protenas anormales se depo sitan primariamente en el espacio extracelular (Captulo 6). El exceso de protenas dentro de las clulas en cantidades suficientes para producir acumulacin visible mo rfolgicamente tiene diversas causas:
38 UNIDAD I Patologa general Defectos en el plegamiento de protenas que pueden subyacer en algunos de estos de psitos en diversas enfermedades no relacionadas76. Las cadenas polipeptdicas nacie ntes de protenas, fabricadas en los ribosomas, finalmente se disponen en hlice a o en lmina p, y la configuracin apropiada de estas disposiciones (plegamiento prote ico) es crtica para la funcin de cada protena y su transporte a las organelas celul ares77. En el proceso de plegamiento, surgen formas intermedias parcialmente ple gadas, y pueden formar agregados intracelulares entre ellas o incluyendo otras p rotenas. Sin embargo, en condiciones normales, estos intermedios se estabilizan m ediante varias chaperonas moleculares, que interactan con las protenas directament e78. Las chaperonas ayudan al plegamiento apropiado y en el transporte a travs de l RE, complejo de Golgi y ms all (Fig. 1-39). Algunas chaperonas se sintetizan con stitutivamente y afectan el trfico normal intracelular de protenas, mientras que o tras estn inducidas por el estrs, tales FIGURA 1-37 Colesterolosis. Macrfagos carga dos de colesterol como el calor (protena de choque calrico, p. ej., hsp70, (clulas espumosas) de un foco de colesterolosis de vescula biliar {flecha). (Cortesa del D r. Matthew Yeh, Universityof Washington, hsp90), y rescatan a las protenas estresad as por el choque del Seattle, WA.) plegamiento errneo. Si el proceso de plegamien to no tiene xito, las chaperonas facilitan la degradacin de la protena daada. Este p roceso degradativo a menudo implica a la ubicuitina (tambin una protena de choque calrico), que se Gotitas de reabsorcin en los tbulos renales proximales que se aade a la protena anormal y la marca para su degradacin ven en las enfermedades renales asociadas con prdida de propor el complejo proteosmico. Existen varios mecanismos por tenas en la orina (proteinuria). En el rion, pequeas cantidades los que el def ecto en el plegamiento proteico puede producir de protenas filtradas a travs de lo s glomrulos se reabsorben acumulacin intracelular o dar lugar a enfermedad. normal mente por pinocitosis en el tbulo proximal. En trastornos con un gran escape prot eico a travs del filtro glomerular, Defectuosos transporte intracelular y secrecin de protenas hay una reabsorcin aumentada de protenas en vesculas. Estas crticas. En el dficit de a,-antitripsina, las mutaciones en la vesculas se runden con los liso somas para producir fagolisosoprotena ralentizan significativamente el plegamient o, mas, que aparecen como gotitas hialinas rosadas dentro del citodando lugar a la construccin de intermediarios parcialplasma de la clula del tbulo (Fig. 1-38). E l proceso es reversimente plegados, que se agregan en el RE del hgado y no ble; s i la proteinuria disminuye, las gotitas de protena se son segregados. La deficien cia resultante de la enzima cirmetabolizan y desaparecen. culante produce enfise ma (Captulo 15). En hfibrosis qustica, la mutacin retrasa la disociacin de una proten a del canal del cloro de una de sus chaperonas, dando lugar a un plegamiento ano rmal y prdida de la funcin (Captulo 10). En la hipercolesterolemia familiar, las mu taciones en los receptores de lipoprotena de baja densidad interfieren con el ple gamiento apropiado de las protenas del receptor (Captulo 5). El estrs del RE induci do por protenas no plegadas y mal plegadas. Las protenas no plegadas o con mal ple gamiento se acumulan en el RE y desencadenan varias respuestas celulares, denomi nadas colectivamente como la respuesta de protena no plegada79's]. La respuesta d e protena no plegada est mediada por varias protenas que residen y se extienden en la membrana del RE. Los dominios luminales de estas protenas captan las alteracio nes en el plegamiento proteico, y los dominios citoplsmicos activan las vas de seal izacin que reducen los niveles de protenas mal plegadas en la clula, aumentando la produccin de FIGURA 1-38 Gotas de reabsorcin proteica en el epitelio tubular renal . (Cortesa del Dr. Helmut Rennke, Department of Pathology, chaperonas y ralentiza ndo la traduccin proteica. ParadBrigham and Women's Hospital, Boston, MA.) jicamen te, la activacin de la respuesta de protena no plegada da lugar tambin a la muerte celular por la activacin de caspasas, particularmente de una caspasa residente en el RE, denominada caspasa-12. As pues, las protenas mal Un segundo caso es la snte sis de cantidades excesivas de plegadas desencadenan, inicialmente, la funcin cit
oproprotena secretora normal, como ocurre en ciertas clulas plastectora de esta re spuesta, pero, si persisten estas protenas mticas comprometidas en la sntesis activ a de inmunoglobuanormales, las funciones citotxicas pro-apoptticas linas. El RE se distiende enormemente produciendo grandes toman el relevo. La agregacin de proten as anormalmeninclusiones homogneas, eosinoflicas, denominadas cuerpos te plegadas, producidas por mutaciones genticas, envejede Russell. cimiento o factores ambien tales desconocidos, actualmen-
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular A PRODUCCIN Y ENSAMBLAJE DE PROTENAS Chaperona mitocondrial (p. ej., Hsp 60) 39 Chaperona (p. ej., Hsp Pptido naciente Protena nitocondrial madura plegada Protenas maduras plegadas RNAm Rlbosomas Chaperona secundarla B REPARACIN DE PROTENAS DAADAS ESTRS (UV, calor, agresin por radicales libres, etc.) Chaperona (p. ej., Hsp) Protena Protenas no funcionales y agregados MUERTE CELULAR Ubicuitina Proteosoma Fragmentos peptdicos degradados x-''J FIGURA 1-39 Mecanismos de plegamiento proteico y papel de las chaperonas. A, las chaperonas, como las protenas de choque calrico (Hsp), protegen a las protenas ple gadas o parcialmente plegadas de la degradacin y guan a las protenas hacia las orga nelas. B, Las chaperonas reparan protenas mal plegadas; cuando este proceso es in eficaz, las protenas son marcadas para degradacin en el proteasoma y si las protena s mal plegadas se acumulan, se desencadena la apoptosis. te se reconoce como una caracterstica de un nmero de especfico de acumulacin. Las ac umulaciones intracelulares de enfermedades neurodegenerativas, incluyendo las en ferprotena descritas antes (gotitas de reabsorcin, cuerpos de Rusmedades de Alzhei mer, Huntington y Parkinson (Captulo sell, hialina alcohlica de Mallory) son ejemp los de depsitos hia28), y posiblemente la diabetes tipo II. La deprivacin de linos intracelulares. glucosa y oxgeno y el estrs como el calor tambin dan La hialina ex tracelular ha sido algo ms difcil de analizar. El lugar a un mal plegamiento de la s protenas y desencadetejido fibroso colgeno en las antiguas cicatrices puede apar ecer nan la respuesta de protena no plegada, culminando en hialinizado, pero el m ecanismofisioqumicosubyacente en que se ' lesin celular y muerte. basa este cambio no est claro. En la hipertensin prolongada y en Agregacin de protenas anormales. La s protenas anorma- la diabetes mellitus, las paredes de las arteriolas, especialm ente les o mal plegadas pueden depositarse en tejidos e interfieen el rion, se to rnan hialinizadas debido a la protena plasmtiren con las funciones normales. Los d epsitos pueden ser ca extravasada y al depsito del material de la membrana basal. intracelulares, extracelulars o ambos, y existe una evidencia acumulada de que lo s agregados pueden producir camGLUCGENO bios patolgicos directa o indirectamente. Ciertas formas de amiloidosis (Captulo 6) entran en la categora de estas El glucgen o es un almacn de energa fcilmente disponible preenfermedades. A esos trastornos se les denomina, a veces, sente en el citoplasma. El exceso de depsitos intracelula
res de proteinopatas o enfermedades por agregacin de protenas.glucgeno se ve en paci entes con anomalas en el metabolismo de la glucosa o del glucgeno. Cualquiera que sea el grupo clnico, las masas de glucgeno aparecen como vacuolas claras dentro de l CAMBIO HIALINO citoplasma. El glucgeno se conserva mejor con fijadores no acuos os; para su localizacin, los tejidos se fijan mejor con alcoEl trmino hialino se r efiere, habitualmente, a una alteracin denhol absoluto. La tincin con carmn Best o el cido perydico de tro de las protenas o en el espacio extracelular que confiere u na apariencia homognea cristalina rosada en los cortes histolgicos Schiff (PAS) im parte un color entre rosa-y-violeta al glucgeno, y de rutina teidos con hematoxili na y eosina. Se utiliza ampliala digestin con diastasa de una seccin paralela ante s de la tinmente como un trmino histolgico descriptivo ms que un marcin sirve como u n control ulterior al hidrolizar el glucgeno. cador especfico de lesin celular. Est e cambio tintorial est proLa diabetes mellitus es el principal ejemplo de un tras torno del ducido por diversas alteraciones y no representa un patrn metabolismo d e la glucosa. En esta enfermedad, el glucgeno se
40 UNIDAD I % Patologa general encuentra en las clulas epiteliales, en las porciones distales de los tubos conto rneados proximales y, a veces, en el asa descendente de Henle, as como en los hep atocitos, clulas |3 de los islotes de Langerhans y en las clulas del msculo cardaco. El glucgeno tambin se acumula dentro de las clulas en un grupo de trastornos estre chamente relacionados, todos ellos genticos, referidos colectivamente como enferm edades de almacenamiento de glucgeno o glucogenosis (Captulo 5). En estas enfermed ades, los defectos enzimticos de la sntesis o fragmentacin del glucgeno dan lugar a una acumulacin masiva, con lesin secundaria y muerte celular. PIGMENTOS tena, sugiriendo que se deriva de la peroxidacin lipdica de los lpidos poliinsaturad os de las membranas subcelulares. La lipofuscina no es lesiva para la clula o sus funciones. Su importancia radica en que es un signo delator de lesin por radical es libres y peroxidacin lipdica. El trmino deriva del latn (fuscus = marrn), por tant o, lpido marrn. En las secciones tisulares aparece como un pigmentofinamentegranul ar amarillo-marrn intracitoplsmico, a menudo perinuclear (Fig. 1-40). Se ve en las clulas sometidas a cambios regresivos lentos y es particularmente prominente en el hgado y en el corazn de los pacientes ancianos o en los pacientes con malnutric in grave o caquexia cancerosa. En el microscopio electrnico, los granulos son muy electrondensos, a menudo tienen estructuras membranosas en su medio y, habitualm ente, son de localizacin perinuclear. Los pigmentos son sustancias coloreadas, algunas de las cuales La melanina, deri vado del griego (melas = negro), es un pigson constituyentes normales de las clul as (p. ej., melanina), mento endgeno, no derivado de la hemoglobina, marrn-negro, mientras que otras son anormales y se coleccionan en las clulas formado cuando la enzima tirosinasa cataliza la oxidacin de la solamente en circunstancias especia les. Los pigmentos pueden ser tirosina a dihidroxifenilalanina en los melanocito s. Se comenta exgenos, provenientes del exterior del cuerpo, o endgenos, sindespus en el Captulo 25. Desde un punto de vista prctico la tetizados dentro del propio c uerpo. melanina es el nico pigmento marrn-negro endgeno. nicaPigmentos exgenos. El ms frecuente de los pigmentos exge- mente el otro que puede considerarse en esta cat egora es el cido nos es el carbn, o polvo de carbn, que es un contaminante areo homog entsico, un pigmento negro que ocurre en pacientes con alcaptonuria, una enfermed ad metablica rara. Aqu, el pigmento ubicuo en la vida urbana. Cuando se inhala, es captado por los se deposita en la piel, tejido conectivo y cartlago, y la pigmen tamacrfagos dentro de los alvolos y, despus, transportado por cin se conoce como ocr onosis (Captulo 5). los canales linfticos hasta los ganglios linfticos regionales e n la regin traqueobronquial. Las acumulaciones de este pigmento La hemosiderina e s un pigmento derivado de la hemoglobina, ennegrecen los tejidos pulmonares (ant racosis) y los ganglios lin- de color amarillo dorado a marrn, granular o cristal ino, en cuya fticos afectados. En los mineros de carbn, los agregados de forma el hierro se almacena en las clulas. El metabolismo del hiepolvo de carbn pueden indu cir una reaccin fibroblstica o rro y la sntesis de ferritina y hemosiderina se cons ideran en detaincluso enfisema y producir, as, una neumopata grave conocida lle en el Captulo 13. Normalmente, el hierro est transportado como neumoconiosis del tra bajador del carbn (Captulo 15). El por protenas de transporte especficas, las transf errinas. En las tatuaje es una forma de pigmentacin exgena localizada en la clulas se almacena asociado a una protena, la apoferritina, para piel. Los pigmentos ino culados son fagocitados por los macrfaformar micelas de ferritina. La ferritina e s un elemento constitugos drmicos, en los que residen por el resto de la vida de la peryente de muchos tipos celulares. Cuando hay un exceso local o sissona embe llecida (a veces con consecuencias embarazosas para el tmico de hierro, la ferriti na forma granulos de hemosiderina, que portador del tatuaje!). Por lo general, l os pigmentos no desencase ven fcilmente con el microscopio ptico (Fig. 1-41). As pu es, denan una respuesta inflamatoria. el pigmento hemosidernico representa agrega dos de micelas de Pigmentos endgenos. La lipofuscina es un pigmento insolu- ferri
tina. En condiciones normales se pueden ver pequeas cantidades de hemosiderina en los fagocitos mononucleares de la ble, conocido tambin como lipocromo, o pigment o de usar y mdula sea, bazo e hgado, todos ellos comprometidos activatirar o pigmen to de la vejez. La lipofuscina est compuesta de mente en la degradacin de los hema tes. polmeros de lpidos y fosfolpidos unidos en complejos con proFIGURA 1-40 Granulos de lipofuscina en un miocito cardaco segn se muestran en A, m icroscopio ptico (depsitos indicados por flechas), y B, microscopio electrnico (ntes e la localizacin perinuclear ntralisosomal).
CAPTULO 1 <Q Adaptaciones celulares, lesin celular y muerte celular 41 FIGURA 1-41 Granulos de hemosiderina en hepatocitos. A, corte con H&E que muestr a pigmentos finamente granulares de color dorado-marrn. B, reaccin de azul de Prus ia especfica para hierro. El exceso de hierro hace que la hemosiderina se acumule en las clulas, bien como un proceso localizado o como un trastorno sistmico. Los excesos locales de hierro y hemosiderina son el resultado de grandes hemorragias o de la mirada de hemorra gias diminutas que acompaan a la congestin vascular intensa. El mejor ejemplo de h emosiderosis localizada es el cardenal comn. Tras la hemorragia local, al princip io el rea est de color rojoazul. Con la lisis de los eritrocitos, la hemoglobina s e transforma finalmente en hemosiderina. Los macrfagos toman parte en este proces o fagocitando los residuos de los hemates y,finalmente,las enzimas lisosomales co nvierten la hemoglobina, a travs de una secuencia de pigmentos, en hemosiderina. Los sucesivos colores por los que atraviesa el cardenal reflejan estas transform aciones. El color original rojo-azul de la hemoglobina se transforma en varios t onos de verde a zul, que reflejan la formacin local de biliverdina (bilis verde), despus bilirrubina (bilis roja) y, a partir de aqu, el componente de hierro de la hemoglobina se deposita en hemosiderina, de color amarillo-dorado. Siempre que haya causas de sobrecarga sistmica de hierro la hemosiderina se depositar en mucho s rganos y tejidos, una situacin denominada hemosiderosis. Se ve con: (1) absorcin aumentada de hierro de la dieta; (2) uso alterado del hierro; (3) anemias hemolti cas, y (4) transfusiones, ya que los hemates transfundidos constituyen una carga exgena de hierro. Estas afecciones se abordan en el Captulo 18. En la mayora de los casos de hemosiderosis sistmica, el pigmento no daa a las clulas parenquimatosas ni deteriora la funcin del rgano. Sin embargo, la acumulacin ms ext remada del hierro, en una enfermedad denominada hemocromatosis, se asocia con dao en el hgado, corazn y pncreas, dando lugar a fibrosis del hgado, insuficiencia carda ca y diabetes mellitus (Captulo 18). La bilirrubina es el pigmento normal ms importante que se encuentra en la bilis. Deriva de la hemoglobina pero no contiene hierro. Su formacin normal y excrecin so n vitales para la salud, y la ictericia es un trastorno clnico frecuente producid o por el exceso de este pigmento en clulas y tejidos. El metabolismo de la bilirr ubina y la ictericia se abordan en el Captulo 18. Calcificacin patolgica La calcificacin patolgica es el depsito anormal en los tejidos de sales de calcio j unto con cantidades menores de hierro, magnesio y otras sales minerales. Es un p roceso frecuente que ocurre en diversas situaciones patolgicas. Existen dos forma s de calcificacin patolgica. Cuando el depsito ocurre ocalmente en tejidos que estn m uriendo, se denomina calcificacin distrfica; ocurre a pesar de niveles sricos norma les de calcio y en ausencia de trastornos en el metabolismo del calcio. En contr aste, el depsito de sales de calcio en tejidos por otra parte normales se conoce como calcificacin metastsica, y casi siempre es el resultado de hipercalcemia secu ndaria a algn trastorno en el metabolismo del calcio. CALCIFICACIN DISTRFICA La cal cificacin distrfica se encuentra en zonas de necrosis, ya sea de tipo por coagulac in, caseosa o por licuefaccin, y en focos de necrosis enzimtica de la grasa. La cal cificacin casi siempre es inevitable en los ateromas de la aterosclerosis avanzad a. Tambin se desarrolla en las vlvulas cardacas envejecidas o daadas, obstaculizando an ms su funcin (Fig. 1-42). Cualquiera que sea el sitio de depsito, las sales de c alcio aparecen macroscpicamente como finos granulos blancos o grumos, a menudo co mo depsitos granulares al tacto. A veces un ganglio linftico tuberculoso adquiere una consistencia ptrea. Morfologa. El pigmento de hierro tiene apariencia de un pigmento granular tosco,
dorado, depositado dentro del citoplasma de las clulas. Cuando la causa bsica es l a rotura localizada de hemates, primero la pigmentacin se encuentra en los fagocit os del rea. En la hemosiderosis sistmica, al principio se encuentra en los fagocit os mononucleares del hgado, mdula sea, bazo y ganglios linfticos, y en macrfagos disp ersos a travs de otros rganos tales como la piel, el pncreas y los rones. Con la acum ulacin progresiva las clulas parenquimatosas de todo el cuerpo (principalmente en el hgado, pncreas, corazn y rganos endocrinos) se pigmentan. El hierro puede visuali zarse en los tejidos con la reaccin histoqumica de azul de Prusia, en la cual el f errocianuro potsico, incoloro, se convierte por el hierro en ferrocianuro frrico, azul oscuro (Fig. 1-416).
42 UNIDAD I ' Patologa general propagacin en la formacin del cristal depende de la concentracin de Ca2+ y P0 4 y l a presencia de inhibidores y otras protenas en el espacio extracelular, como las protenas matriciales del tejido conectivo. Aunque la calcificacin distrfica puede s er simplemente un signo delator de una lesin celular previa, a menudo es la causa de disfuncin del rgano. Tal es el caso en la enfermedad valvular calcificada y en la aterosclerosis, como se aclarar en la exposicin ulterior sobre estas enfermeda des. CALCIFICACIN METASTSICA La calcificacin metastsica puede ocurrir en tejidos normales siempre que haya hipe rcalcemia. La hipercalcemia tambin acenta la calcificacin distrfica. Hay cuatro caus as principales de hipercalcemia: (1) secrecin aumentada de hormona paratiroidea ( PTH) con resorcin sea subsiguiente, como en el hiperparatiroidismo debido a tumore s paratiroideos y la secrecin ectpica de protena relacionada con la PTH por tumores malignos (Captulo 7); (2) destruccin del tejido seo, que ocurre en los tumores pri marios de mdula sea (p. ej., mieloma mltiple, leucemia) o metstasis esquelticas difus as (p. ej., cncer de mama), recambio seo acelerado (p. ej., enfermedad de Paget), o inmovilizacin; (3) trastornos relacionados con la vitamina D, incluyendo intoxi cacin por vitamina D, sarcoidosis (en la cual los macrfagos activan un precursor d e la vitamina D), e hipercalcemia idioptica de la infancia (sndrome de Williams), caracterizado por sensibilidad anormal a la vitamina D, y (4) insuficiencia rena l, que produce retencin de fosfato, dando lugar a hiperparatiroidismo secundario. Las causas menos comunes incluyen intoxicacin por aluminio, que ocurre en pacien tes en dilisis renal crnica, y en el sndrome de leche-alcalinos, que se debe a una ingestin excesiva de calcio y anticidos absorbibles como la leche o el carbonato c alcico. FIGURA 1-42 Vista desde arriba de la vlvula artica cerrada en un corazn con estenos is artica calcificada. Las cspides semilunares estn engrosadas y fibrticas. Detrs de cada cspide se ven masas irregulares y calcificacin distrfica apilada. La calcificacin metastsica puede ocurrir ampliamente en todo el cuerpo pero afecta , principalmente, a los tejidos intersticiales de la mucosa gstrica, rones, pulmone s, arterias sistmicas y venas pulmonares. Aunque completamente diferentes en su l ocalizacin, todos estos tejidos pierden cido y, por lo tanto, tienen un compartime nto alcalino interno que los predispone a la calcificacin metastsica. En todos eso s sitios, las sales de calcio se asemejan morfolgicamente a las descritas en la c alcificacin disPatogenia. En la patognesis de la calcificacin distrfica, la va trfica. Por tanto, pueden ocurrir como depsitos amorfos no final comn es la formacin de mi neral de fosfato calcico cristali- cristalinos y, otras veces, como cristales de hidroxiapatita. no en forma de apatita, similar a la hidroxiapatita del hueso. El Habitualmente, las sales minerales no producen disfuncin clproceso tiene dos fa ses principales: iniciacin (o nucleacin) y nica pero, a veces, la afectacin masiva de los pulmones produce propagacin; ambas ocurren intracelular y extracelularment e. La radiografas llamativas y deficiencias respiratorias. Los depsitos iniciacin d e la calcificacin intracelular ocurre en las mitocon- masivos en el rion (nefrocal cinosis) pueden producir, con el drias de las clulas muertas o moribundas que acu mulan calcio. tiempo, dao renal (Captulo 20). Entre los iniciadores de la calcific acin distrfica extracelular se incluyen los fosfolpidos, que se encuentran en vescul as ligadas a Envejecimiento celular la membrana, con un dimetro de aproximadament e 200 nm; en el cartlago y en el hueso se conocen como vesculas matriciales y, Pro bablemente Shakespeare fuera el mejor en caracterizar el en la calcificacin patolg ica, derivan de clulas degeneradas o envejecimiento en su elegante descripcin de l as siete edades del envejecidas. Se piensa que el calcio se concentra en estas v escuhombre. Comienza en el momento de la concepcin, implica la las por un proceso de calcificacin facilitado por la membrana diferenciacin y maduracin del organismo
y sus clulas, en de varias etapas: (1) el ion calcio se une a los fosfolpidos prea lgn punto variable del tiempo da lugar a la prdida progresiva sentes en la membran a de la vescula; (2) las fosfatasas asociadas de la capacidad funcional caracterst ica de la senectud, y acaba en a la membrana generan grupos fosfato, que se unen al calcio; la muerte. (3) se repite el ciclo de unin entre calcio y fosfato, ele vando las Con la edad, hay alteracionesfisiolgicasy estructurales en casi concent raciones locales y produciendo un depsito cerca de todos los rganos y sistemas. El envejecimiento en los individuos la membrana, y (4) ocurre un cambio estructura l en la disposiest afectado en gran medida por factores genticos, la dieta, concin de los grupos de calcio y fosfato, generando un microcrisdiciones sociales y la aparicin de enfermedades relacionadas con tal que, entonces, puede propagarse y p erforar la membrana. La la edad, como la aterosclerosis, la diabetes y la artrit is. Adems, Morfologa. Histolgicamente, con la tincin habitual de hematoxilina y eosina las sal es de calcio tienen una apariencia basoflica granular amorfa, a veces agrupada. P ueden ser intracelulares, extracelulares, o con ambas localizaciones. Con el tra nscurso del tiempo puede formarse hueso heterotpico en los focos de calcificacin. A veces, las clulas necrticas aisladas pueden constituir el inicio para el depsito de minerales. La adquisicin progresiva de capas externas puede crear configuracio nes laminares, denominadas cuerpos de psamoma por su semejanza con granulos de a rena. Algunos tipos de cncer papilar (p. ej., de tiroides) tienen propensin a desa rrollar cuerpos de psamoma. Cuando las sales de calcio y hierro se juntan alrede dor de largas espculas delgadas de asbestos en el pulmn, emergen concreciones extr aas que crean formas exticas, arrosariadas, en aspecto de pesa.
CAPTULO 1 % Adaptaciones celulares, lesin celular y muerte celular 43 existe buena evidencia de que las alteraciones celulares inducidas 20 por la eda d son componentes importantes del envejecimiento del organismo. Exponemos aqu el envejecimiento celular porque Recin nacido podra representar la acumulacin progresi va con los aos de 15 lesin subletal que podra dar lugar a la muerte celular o, al m enos, a una capacidad disminuida de la clula para responder a la lesin. 3 El envej ecimiento celular es el resultado de la declinacin progre- 10 siva en la capacida dproliferativa y la duracin de la vida de las clulas, y de los efectos de la expos icin continuada a influencias exge-D nas que dan lugar a la acumulacin progresiva d e dao celular y O (Z 5LU molecular (Fig. 1-43). Estos procesos se revisan a conti nuacin. Cambios estructurales y bioqumicos del envejecimiento celular. Varias func iones celulares declinan progresivamente con la edad. La fosforilacin oxidativa p or las mitocondrias est redu-i 1i * i i cida, as como la sntesis de cidos nucleicos as protenas estruc80 10 20 30 40 50 60 70 turales y enzimticas, los receptores cel ulares y los factores de NIVEL DE DUPLICACIN CELULAR transcripcin. Las clulas senes centes tienen una capacidad disFIGURA 1-44 Duplicaciones finitas de la poblacin d e fibroblasminuida para captar nutrientes y reparar el dao cromosmico. tos humanos primarios tomados de un recin nacido, una persoLas alteraciones morfolgicas en la s clulas envejecidas incluyen na de 100 aos y un paciente de 20 aos con sndrome de W erner. ncleos irregulares y anormalmente lobulados, mitocondrias La capacidad de las clulas para crecer hasta una monocapa confluente disminuye con los niveles cr ecientes de duplicacin de la pleomrficas vacuoladas, retculo endoplsmico disminuido y poblacin. (De Dice JF: Cellular and molecular mechanisms of aparato de Golgi di storsionado. A la vez, hay una acumulacin aging. Physiol Rev 73:150, 1993.) conti nuada del pigmento lipofuscina que, como hemos visto, representa un producto de peroxidacin lipdica y evidencia de dao oxidativo; productos terminales de la glucac in avanzada, que son el resultado de la glucosilacin no enzimtica y son capaces de reducida in vitro. Tras un nmero determinado de divisiones, establecer enlaces cr uzados entre protenas adyacentes, y la acutodas las clulas se paran en un estado t erminal sin divisin, mulacin de protenas anormalmente plegadas. El papel del dao con ocido como senescencia celular. Durante el envejecimiento oxidativo se comenta ms adelante. Los productos terminales de celular ocurren muchos cambios en la expr esin del gen, pero una la glucacin avanzada son importantes en la patogenia de la diacuestin clave es cules son las causas y cules los efectos de la betes mellitus y se exponen en el Captulo 24, pero tambin puesenescencia celular83. Por ejemplo, a lgunas de las protenas que den participar en el envejecimiento. Por ejemplo, la g lucosilacin inhiben la progresin del ciclo de crecimiento celular (como se de las protenas del cristalino relacionada con la edad puede subdetalla en el Captulo 7) -tales como los productos de los genes yacer en las cataratas seniles. La natura leza de las protenas anorinhibidores de la cinasa dependiente de ciclina (p. ej., p21)- se malmente plegadas se coment antes en este captulo. sobreexpresan en las clulas senescentes. Senescencia replicativa. El concepto de que las clulas tienen El cmo las clulas en divisin pueden contar sus divisiones es una capacidad limitada de replicacin se desarroll a partir de un el objeto de una intensa investigacin. U n mecanismo probable modelo experimental simple de envejecimiento. Los fibroblas tos es el que en cada divisin celular existe una replicacin incomplehumanos normal es, cuando se colocan en un cultivo tisular, tieta de los extremos de los cromos omas (acortamiento del telmero), nen un potencial de divisin limitado82. Las clulas de los nios que finalmente da lugar a la detencin del ciclo celular. Los telmesufr en ms ciclos de replicacin que las clulas de personas mayoros son secuencias cortas repetidas de DNA (TTAGGG) presentes res (Fig. 1-44). En contraste, las clulas de los pacientes con snen el extremo lineal de los cromosomas y son importantes par a drome de Werner, una enfermedad rara caracterizada por enveje- asegurar la rep licacin completa de las terminales cromosmicas cimiento prematuro, tienen una dura cin de vida marcadamente y proteger dichas terminales de la fusin y degradacin84'85 .
FACTORES GENTICOS Defectos en la reparacin Varias del DNA anomalas genticas (p. ej., vadellGF-1) FACTORES AMBIENTALES Agresiones ambientales T Actividad reducida Dao mediado del proteosoma por radicales libres Acumulacin de mutaciones Sealizacin celular anormal Acumulacin de protenas y organelas celulares daadas I FIGURA 1-43 Mecanismos de envejecimiento celular. Los factores genticos y las agr esiones ambientales se combinan para producir las anomalas celulares caracterstica s del envejecimiento. Senescencia replicativa I Capacidad reducida para producir nuevas clulas 1 I ENVEJECIMIENTO CELULAR
44 UNIDAD I % Patologa general Cuando las clulas somticas se replican, una pequPa seccin del telmero no se duplica, y los telmeros se acortan progresivamente. Segn los telmeros se van acortando, los extremos de los cromosomas no pueden protegerse y se ven como DNA roto, lo que s ealiza la detencin del ciclo celular. Las longitudes de los telmeros se mantienen n ormales por adicin de nucletido mediada por una enzima denominada telomerasa. La t elomerasa es un complejo especializado de RNA-protena que utiliza su propio RNA c omo un molde para aadir nucletidos en los extremos de los cromosomas (Fig. 1-45). La actividad de la telomerasa est reprimida por protenas reguladoras que restringe n la elongacin del telmero, proporcionando as un mecanismo sensor de longitud. La a ctividad telomerasa se expresa en las clulas germinales y est presente en niveles bajos en las clulas madre, pero est habitualmente ausente en la mayora de los tejid os somticos. Por lo tanto, segn envejecen las clulas, sus telmeros se hacen ms cortos y salen del ciclo celular dando lugar a la incapacidad de generar nuevas clulas para reemplazar las daadas. A la inversa, en las ~Hebra ori inal clulas cancerosas inmortales, la telomerasa se reactiva y los telmeros no estn acor tados, sugiriendo que la elongacin del telmero podra ser un paso importante -posibl emente esencialen la formacin del tumor85. Sin embargo, a pesar de tales atractiv as observaciones, an queda por establecer por completo la relacin entre la activid ad telomerasa y la longitud telomrica con el envejecimiento y el cncer86. Genes qu e influyen sobre el proceso de envejecimiento. Los estudios con Drosopha, C. eleg ans y ratones estn conduciendo al descubrimiento de genes que influyen sobre el p roceso de envejecimiento87. Un grupo interesante de genes implica la va de insuli na/factor de crecimiento insulnico-1. La disminucin de sealizacin a travs del recepto r de IGF-1 como resultado de una disminucin de la ingestin calrica, o mutaciones en el receptor dan lugar a una prolongacin de la vida de C. elegans. Las seales post eriores al receptor de IGF-1 implican varias cinasas y pueden dar lugar al silen ciamiento de genes concretos, favoreciendo as el envejecimiento. Los anlisis de hu manos con envejecimiento preI I I I I I I I I II I I I I I TTAGGGTTAGGGTTAG AATCCCA 9 L-L51 Hebra de nueva sntesis (incompleta) Unin de telomerasa Molde de RNA AATCCCA mmmmmr Clulas germinales Extensin del extremo 3' X^s^s^.GGGTTAGGGTUUGTAG AATCCCA o cE E o un B "o x> o
\. Activacin de la telomer asa Clula cancerosa . * * * Detencin \ del crecimiento *, La polimerasa de DNA completa la hebra retrasada \ B Divisiones celulares FIGURA 1-45 Papel de los t e l m e r o s y de la t e l o m e r a s a en la senes cencia replicativa de las clulas. A, la t e l o m e r a s a d i r i g e la sntesis de D N A d e p e n d i e n t e del m o l d e de RNA, en la que los n u c l e t i d o s se aaden a una hebra al final de un c r o m o s o m a . La hebra retrasad a se c o m p l e t a , p r e s u m i b l e m e n t e , m e d i a n t e la p o l i m e r a s a a de DNA. La secuencia de RNA en la t e l o m e r a s a es diferen te en las diversas especies ( M o d i f i c a d o de A l b e r t s BR, et al: Mo lecular B i o l o g y of the Cell, 2002, Garland Science, N e w York.) 6, hiptesi s t e l m e r o - t e l o m e r a s a y capacidad proliferativa. La l o n g i t u d del t e l m e r o se c o m p a r a con el n m e r o de divisiones celulares. En las clulas somticas n o r m a l e s , no hay actividad telomerasa y los t e l m e r o s se acortan p r o g r e s i v a m e n t e al a u m e n t a r las divisi ones celulares hasta que ocurre la d e t e n cin del c r e c i m i e n t o o sene scencia. Las clulas g e r m i n a l e s y las clulas m a d r e c o n t i e n e n t e l o m e r a s a activa, pero s o l a m e n t e las clulas g e r m i n a l e s presentan suficientes niveles de la enzima para estabilizar c o m p l e t a m e n t e la l o n g i t u d del t e l m e r o . La activacin de la t e l o merasa en clulas cancerosas inactiva el reloj t e l o m r i c o que limita la capacidad pr oliferativa de las clulas somticas n o r m a l e s . ( M o d i f i c a d o y r e d i s e a d o c o n p e r m i s o de Holt SE, et a l . : R e f i n i n g t h e t e l o m e r - t e l o m e r a s e h y p o t h e s i s of a g i n g a n d cncer. N a t u r e B i o t e c h 14:836,1996. C o p y r i g h t 1996, M a c m i l l a n Magazines Limited.)
CAPTULO 1 # Adaptaciones celulares, lesin celular y muerte celular 45 maturo tambin estn estableciendo el concepto fundamental de que el envejecimiento no es un proceso al azar sino que est regulado por genes, receptores y seales espe cficos88. Acumulacin de dao metablico y gentico. Adems de la importancia del tiempo co mo reloj gentico, la duracin de la vida celular tambin puede estar determinada por el equilibrio entre el dao celular resultante de acontecimientos metablicos que oc urren dentro de la clula y las respuestas moleculares opuestas que pueden reparar el dao. Por lo general, los animales pequeos tienen duraciones de vida ms cortas y tasas metablicas ms rpidas, sugiriendo que la duracin de la vida de las especies es t limitada por el consumo metablico total fijo a lo largo de la vida89. Un grupo d e productos de un metabolismo normal son las formas de oxgeno reactivo. Como hemo s visto, estos subproductos de la fosforilacin oxidativa producen modificaciones covalentes de protenas, lpidos y cidos nucleicos. La cantidad de dao oxidativo, que aumenta al envejecer el organismo, puede ser un componente importante de la sene scencia, y la acumulacin de lipofuscina en las clulas envejecidas se considera un signo de tal dao. Con esta propuesta son coherentes las siguientes observaciones: (1) la variacin en la longevidad en diferentes especies se correlaciona inversam ente con las tasas de generacin del radical anin superxido en las mitocondrias, y ( 2) la sobreexpresin de las enzimas antioxidantes superxido dismutasa (SOD) y catal asa prolonga la duracin de la vida en las formas transgnicas de Drosophila. As pues , parte del mecanismo de control de la cronologa del envejecimiento puede ser el dao acumulado que se genera por subproductos txicos del metabolismo, tales como ra dicales de oxgeno. El aumento del dao oxidativo puede ser el resultado de exposici ones ambientales repetidas a agentes tales como la radiacin ionizante, la reduccin progresiva de los mecanismos de defensa antioxidante (p. ej., vitamina E, gluta tin peroxidasa) o ambas cosas. Varias respuestas protectoras contrarrestan el dao progresivo en las clulas, y una importante es el reconocimiento y reparacin del DN A daado90. Aunque la mayor parte del dao del DNA se repara mediante las enzimas en dgenas reparadoras del DNA, algo persiste y se acumula a medida que las clulas env ejecen. Varias lneas de evidencia apuntan a la importancia de la reparacin del DNA en el proceso de envejecimiento. Los pacientes con sndrome de Werner muestran en vejecimiento prematuro, y el producto de los genes defectuosos es una helicasa d e DNA -una protena implicada en la replicacin y reparacin del DNA y en otras funcio nes que requieren el desenrollamiento del DNA91-. Un defecto en esta enzima prod uce una acumulacin rpida de dao cromosmico que simula la lesin que normalmente se acu mula durante el envejecimiento celular. La inestabilidad gentica en clulas somticas tambin es caracterstica de otros trastornos en los que los pacientes exhiben algu na de las manifestaciones del envejecimiento acelerado, como la ataxia-telangiec tasia, en la cual el gen mutado codifica una protena implicada en la reparacin de las roturas de la doble cadena del DNA (Captulo 7). Los estudios de los mutantes de levaduras asexuadas y C. elegans muestran que la duracin de la vida se prolong a si estn aumentadas las respuestas al dao del DNA. As pues, el equilibrio entre el dao metablico acumulativo y la respuesta a este dao podra determinar la velocidad a la cual envejecemos. En este escenario el envejecimiento puede retrasarse dismi nuyendo la acumulacin del dao o aumentando la respuesta al mismo. Adems del DNA daad o, tambin las organelas celulares daadas se acumulan al envejecer las clulas. En pa rte esto puede ser el resultado de una funcin declinante del proteosoma, la maquinaria proteoltica que sirve para eliminar protenas intracelulares anormales o indeseadas92. En conclusin, debera quedar claro que las diversas formas de deteri oros celulares y adaptaciones descritas en este captulo cubren un espectro amplio , que se extiende desde adaptaciones en el tamao celular, crecimiento y funcin, pa sando por las formas reversibles e irreversibles de la lesin celular aguda, por e l tipo de muerte celular regulada representada por la apoptosis, por las alterac iones patolgicas en las organelas celulares y hasta las formas menos ominosas de acumulacin intracelular, incluyendo las pigmentaciones. A lo largo de este libro se hace referencia a todas estas alteraciones porque toda lesin orgnica y, en ltima
instancia, todas las enfermedades clnicas se originan de alteraciones en la estr uctura y funcin celulares. BIBLIOGRAFA 1. Majno G: The Healing Hand: Man and Wound in the Ancient World. Cambridge: Har vard University Press, 1975, p 43. 2. Taub R: Transcriptional control of liver r egeneration. FASEB J 10:413,1997. 3. Thorgeirsson SS: Hepatic stem cells in live r regeneration. FASEB J 10:1249, 1996. 4. Forbes S, et al: Hepatic stem cells. J Pathol 197:510, 2002. 5. Korbling M, Estrovz Z: Adult stem cells for tissue rep air: a new therapeutic concept? New Eng J Med 349:570, 2003. 6. Anversa P, Nadal -Ginard B: Myocyte renewal and ventricular remodeling. Nature 415:240, 2002. 7. Molkentin JD, Dorn GW: ytoplasmic signaling pathways that reglate cardiac hypertro phy. Annu Rev Physiol 63:391, 2001. 8. MacLellan WR, Schneider MD: Genetic disse ction of cardiac growth control pathways. Annu Rev Physiol 62:289, 2000. 9. Sait o Y, et al: Augmented expression of atrial natriuretic polypeptide gene in ventr icle of human failing heart. J Clin Invest 83:298, 1989. 10. Kozma SC, Thomas G: Regulation of cell size in growth, development and human disease: PI3K, PKB and S6K. Bioessays 24:65, 2002. 11. Anversa P, et al: Myocyte death in heart failur e. Curr Opin Cardiol 11:245,1996. 12. Glickman MH, Ciechanover A: The ubiquitinproteasome proteolytic pathway: destruction for the sake of construction. Physio l Rev 82:373, 2002. 13. Lugo M, Putong PB: Metaplasia: an overview. Arch Pathol Lab Med 108:185, 1984. 14. Tosh D, Slack JM: How cells change their phenotype. N at Rev Mol Cell Biol 3:187, 2002. 15. Reddi HA: BMPs: actions in flesh and bone. Nat Med 3:837, 1997. 16. Ross SA, McCaffrey PJ, Drager UC, DeLuca LM: Retinoids in embryonal development. Physiol Rev 80:1021, 2000. 17. Newmeyer DD, FergusonMiller S: Mitochondria: releasing power for life and unleashing the machtneries of death. Cell 112:481, 2003. 18. Trump BF, Berezesky I: The reactions of cells to lethal injury: oncosis and necrosisthe role of calcium. In Lockshin RA (ed): W hen Cells DieA Comprehensive Evaluation of Apoptosis and Programmed Cell Death. N ew York: Wiley-Liss, 1998, p 57. 19. Orrenius S, Zhivotovsky B, Nicotera P: Regu lation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552, 2 003. 20. Paschen W: Role of calcium in neuronal cell injury: which subcellular c ompartment is involved? Brain Res Bull 53:409, 2000. 21. Droge W: Free radicis in the physiological control of cell function. Physiol Rev 82:47, 2002. 22. Hensle y K, Robinson KA, Gabbita SP, Salsman S, Floyd RA: Reactive oxygen species, cell signaling, and cell injury. Free Radie Biol Med 28:1456,2000. 23. Salvemini D, Cuzzocrea S: Superoxide, superoxide dismutase and ischemic injury. Curr Opin Inv estig Drugs 3:886, 2002. 24. Li C, Jackson RM: Reactive species mechanisms of ce llular hypoxiareoxygenation injury. Am J Physiol Cell Physiol 282:C227, 2002. 25 . Mitch WE, Goldberg AE: Mechanisms of muscle wasting: the role of the ubiquitin -proteosome pathway. N Engl J Med 335:1897, 1996. 26. Kim JS, He L, Lemasters JJ : Mitochondrial permeability transition: a common pathway to necrosis and apopto sis. Biochem Biophys Res Commun 304:463, 2003. 27. Trump BF, et al: Cell injury and cell death: apoptosis, oncosis, and necrosis. In Acosta D (ed): Cardiovascul ar Toxicology, 3rd ed. London and New York: Taylor & Francis, 2001, p 105.
46 UNIDAD I ' Patologa general 28. Sheridan AM, Bonventre JV: Cell biology and molecular mechanisms of 29. 30. 31. 32. 33. 34. 35. injury in ischemic acute renal failure. Curr Opin Ne phral Hypertens 9:427, 2000. Hou ST, McManus JP: Molecular mechanisms of cerebra l ischemiainduced neuronal death. Int Rev Cytol 221:93, 2002. Daemen MARC, De Vr ies B, Buurman WA: Apoptosis and inflammation in renal reperfusion injury. Trans plantation 73:1693, 2002. Anaya-Prado R, et al: Ischemia/reperfusion injury. J S urg Res 105:248, 2002. Kaminski KA, et al: Oxidative stress and neutrophil activ ation the two keystones of ischemia/reperfusion injury. Int J Cardiol 86:41, 200 2. Thiagarajan RR, et al: The role of leukocyte and endothelial adhesin molecules in ischemia-reperfusion injury. Thromb Haemost 78:310, 1997. Riedemann NC, Ward PA: Complement in ischemia reperfusion injury. Am J Pathol 162:363, 2003. Weise r MR, et al: Reperfusion injury of ischemic skeletal muscle is mediated by natur al antibody and complement. J Exp Med 183:2343, 1996. Snyder JW: Mechanisms of t oxic cell injury. Clin Lab Med 10:311, 1990. Coon MJ, et al: Cytochrome P450: pe roxidative reactions of diversozymes. FASEB J 10:428, 1996. Gonzlez FJ: The use o f gene knockout mice to unravel the mechanisms of toxicity and chemical carcinog enesis. Toxicol Lett 120:199, 2001. Plaa GL: Chlorinated methanes and liver inju ry: highlights of the past 50 years. Annu Rev Pharmacol Toxicol 40:42, 2000. Jae schke H, et al: Mechanisms of hepatotoxicity. Toxicol Sci 65:166, 2002. Cohn SD, Khairallah EA: Selective protein arylation and acetaminophen-induced hepatotoxic ity. Drug Metab Rev 29:59, 1997. Kerr JF, et al: Apoptosis: a basic biological p henomenon with wideranging implications in tissue kinetics. Br J Cncer 26:239, 19 72. Metzstein MM, Stanfield GM, Horvitz HR: Genetics of programmed cell death in C. elegans: past, present and future. Trends Genet 14:410, 1998. Wyllie AH: Apo ptosis: an overview. Br Med Bull 53:451, 1997. Strasser A, O'Connor L, Dixit VM: Apoptosis signaling. Annu Rev Biochem 69:217, 2000. Vaux D, Silke J: Mammalian mitochondrial IAP-binding proteins. Biochem Biophys Res Commun 203:449, 2003. Mc Carthy NJ, Evan GI: Methods for detecting and quantifying apoptosis. Curr Top De v Biol 36:259, 1998. Hanayama R, et al: Identification of a factor that links ap optotic cells to phagocytes. Nature 417:182, 2002. Savill J, Fadok V: Corpse cle aring defines the meaning of cell death. Nature 407:784, 2000. Vaux DL, Strasser A: The molecular biology of apoptosis. Proc Nati Acad Sci U S A 93:2239, 1996. Wallach D, et al: Tumor necrosis factor receptor and Fas signaling mechanisms. A nnu Rev Immunol 17:331, 1999. Thome M, Tschopp J: Regulation of lymphocyte proli feration and death by FLIP. Nat Rev Immunol 1:50, 2001. Van Blitterswijk WJ, et al: Ceramide: second messenger or modulator of membrane structure and dynamics? Biochem J 369:199, 2003. Scorrano L, Korsmeyer SJ: Mechanisms of cytochrome relas e by proapoptotic BCL-2 family members. Biochem Biophys Res Commun 304:437, 2003 . Ravagnan L, Roumier T, Kroemer G: Mitochondria, the killer organdes and their w eapons. J Cell Physiol 192:131, 2002. Cory S, Adams JM: The Bcl2 family: regulat ors of the cellular life-ordeath switch. Nature Rev Cncer 2:647, 2002. Reed JC: C ytochrome c: can't live with it can't live without it. Cell 91:559, 1997. Salves en GS, Duckett CS: IAP proteins: blocking the road to death's door. Nature Rev M ol Cell Biol 3:401, 2002. Joza N, Kroemer G, Penninger JM: Genetic analysis of t he mammalian cell death machinery. Trends Genet 18:142, 2002. 60. Salvesen GS, Dixit VM: Caspases: intracellular signaling by proteolysis. Cell 91:443, 1997. 61. Ravichandran KS: "Recruitment signis" from apoptotic cells : invitation to a quiet meal. Cell 113:817, 2003. 62. Rathmell JC, Thompson CB: Pathways of apoptosis in lymphocyte development, homeostasis, and disease. Cell 109:S97, 2002. 63. Vousden KH, Lu X: Live or let die: the cell's response to p53 . Nature Rev Cncer 2:594, 2002. 64. Siegel RM, et al: The multifaceted role of Fa s signaling in immune cell homeostasis and autoimmunity. Nat Immunol 1:469, 2000 . 65. Locksley RM, Killeen N, Lenardo MJ: The TNF and TNF receptor superfamilies : integrating mammalian biology. Cell 104:487, 2001. 66. Russell JH, Ley TJ: Lym phocyte-mediated cytotoxicity. Annu Rev Immunol 20:323, 2002. 67. Webb SJ, et al
: Apoptosis. An overview of the process and its relevance in disease. Adv Pharma col 41:1, 1997. 68. Dunn WA: Studies on the mechanisms of autophagy. J Cell Biol 110:1923, 1990. 69. Klionsky DJ, Emr SD: Autophagy as a regulated pathway of ce llular degradation. Science 290:1717, 2000. 70. Di Mauro S, Schon EA: Mitochondr ial respiratory-chain diseases. New Engl J Med 348:2656, 2003. 71. Mermall V, et al: Unconventional myosins in cell movement, membrane traffic and signal transd uction. Science 279:527, 1998. 72. Fuchs E, Cleveland DW: A structural scaffoldi ng of intermediate filaments in health and disease. Science 279:514, 1998. 73. D enk H, Stumptner C, Zatloukal K: Mallory bodies revisited. J Hepatol 32:689, 200 0. 74. Snapper SB, Rosen FS: The Wiskott-Aldrich syndrome protein (WASP): roles in signaling and cytoskeletal organization. Annu Rev Immunol 17:905, 1999. 75. L ee RJ: Fatty change and steftohepatitis. In Lee RJ (ed): Diagnostic Liver Patholo gy. St. Louis: Mosby-Year Book, 1994, p 167. 76. Soto C: Protein misfolding and disease; protein refolding and therapy. FEBS Lett 498:204, 2001. 77. Horwich A: Protein aggregation in disease: a role for folding intermediates forming specifi c multimeric interactions. J Clin Invest 110:1221, 2002. 78. Hartl FU, Hayer-Har tl M: Molecular chaperones in the cytosol: from nascent chain to folded protein. Science 295:1852, 2002. 79. Kaufman RJ: Orchestrating the unfolded protein resp onse in health and disease. J Clin Invest 110:1389, 2002. 80. Patil C, Walter P: Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfo lded protein response in yeast and mammals. Curr Opin Cell Biol 13:349, 2001. 81 . Ma Y, Hendershot LM: The unfolding tale of the unfolded protein response. Cell 107:827,2001. 82. Hayflick L, Moorhead PS: The serial cultivation of human dipl oid cell strains. Exp Cell Res 25:585, 1961. 83. Smith JR, Pereira-Smith OM: Rep licative senescence: implications for in vivo aging and tumor suppression. Scien ce 273:63, 1996. 84. Blackburn EH: Switching and signaling at the telomere. Cell 106:661, 2001. 85. Wong JMY, Collins K: Telomere maintenance and disease. Lance t 362:983, 2003. 86. Stewart SA, Weinberg RA: Senescence: does it all happen at the ends? Oncogene 21:627, 2002. 87. Guarente L, Kenyon C: Genetic pathways that reglate ageing in model organisms. Nature 408:255, 2000. 88. Martin GM, Oshima J : Lessons from human progeroid syndromes. Nature 408:263, 2000. 89. Finkel T, Ho lbrook NJ: Oxidants, oxidative stress, and the biology of ageing. Nature 408:239 , 2000. 90. Gilchrest BA, Bohr VA: Aging processes, DNA damage, and repair. FASE B J 11:322,1997. 91. Bohr VA: Human premature aging syndromes and genomic instab ility. Mech Ageing Dev 123:987, 2002. 92. Carrard G, et al: Impairment of protea some structure and function in aging. Int J Biochem Cell Biol 34:1461, 2002. 36. 37. 38. 39. 40. 41. 42. 43. 44. 45. 46. 47. 48. 49. 50. 51. 52. 53. 54. 55. 56. 57. 58. 59.
Vous aimerez peut-être aussi
- El Poder Del Pensamiento PositivoDocument103 pagesEl Poder Del Pensamiento PositivoJosue Amador88% (8)
- .La Nueva Medicina GermanicaDocument60 pages.La Nueva Medicina Germanicabeobay100% (1)
- Programa de Pausas ActivasDocument6 pagesPrograma de Pausas ActivasJudy TorresPas encore d'évaluation
- Test Habilidades GerencialesDocument11 pagesTest Habilidades Gerencialesalguien100% (3)
- MANUALDocument104 pagesMANUALelizabeth baltazar moreno100% (2)
- 1562-Manual Básico de Historia Del Arte (2018) PDFDocument200 pages1562-Manual Básico de Historia Del Arte (2018) PDFPablo Martin RamosPas encore d'évaluation
- Historia Universal Asimov - La Tierra de CanaanDocument186 pagesHistoria Universal Asimov - La Tierra de CanaanGeorgedelaselva100% (13)
- La Inhibición de La Acción Henri LaboritDocument12 pagesLa Inhibición de La Acción Henri LaboritRayGerasPas encore d'évaluation
- Michael Eigen, Wilfred Bion y El Contacto Con Las Profundidades Juan José Martínez IbáñezDocument13 pagesMichael Eigen, Wilfred Bion y El Contacto Con Las Profundidades Juan José Martínez IbáñezJesús N. MorenoPas encore d'évaluation
- Carta de Invitación Alianza CaribeDocument1 pageCarta de Invitación Alianza CaribeHernán Reales VegaPas encore d'évaluation
- Consulta El Acervo 1Document1 pageConsulta El Acervo 1Hernán Reales VegaPas encore d'évaluation
- Crisis, Diálogos Y Desafíos en El Gran Caribe Xii Seminario Internacional de Estudios Del CaribeDocument529 pagesCrisis, Diálogos Y Desafíos en El Gran Caribe Xii Seminario Internacional de Estudios Del CaribeHernán Reales VegaPas encore d'évaluation
- DE LA HOZ Empresas de Vapores en El Caribe Colombiano PDFDocument35 pagesDE LA HOZ Empresas de Vapores en El Caribe Colombiano PDFCarlos Eduardo Valencia VillaPas encore d'évaluation
- Terminos Arq. AbaluartadaDocument4 pagesTerminos Arq. AbaluartadaHernán Reales VegaPas encore d'évaluation
- 2173-Texto Del Artículo-7421-1-10-20101028Document21 pages2173-Texto Del Artículo-7421-1-10-20101028Hernán Reales VegaPas encore d'évaluation
- Terminos Arq. Abaluartada PDFDocument19 pagesTerminos Arq. Abaluartada PDFFrancisco BadasPas encore d'évaluation
- Rev Ilustrada Febrero 1899Document16 pagesRev Ilustrada Febrero 1899Hernán Reales VegaPas encore d'évaluation
- Estructura de La Propiedad de Predios en PDFDocument247 pagesEstructura de La Propiedad de Predios en PDFHernán Reales VegaPas encore d'évaluation
- Asimov, Isaac - Guia de La Biblia - Antiguo TestamentoDocument2 pagesAsimov, Isaac - Guia de La Biblia - Antiguo TestamentoHernán Reales VegaPas encore d'évaluation
- 06 Inductores de ValorDocument14 pages06 Inductores de ValoralexmorenoasuarPas encore d'évaluation
- Inglés Curso 4 Colombo AmericanoDocument3 pagesInglés Curso 4 Colombo AmericanoHernán Reales VegaPas encore d'évaluation
- Programa Hist. Latinoamericana I - Prof. Katia PadillaDocument9 pagesPrograma Hist. Latinoamericana I - Prof. Katia PadillaHernán Reales VegaPas encore d'évaluation
- COTIZACION MariferhtDocument1 pageCOTIZACION MariferhtHernán Reales VegaPas encore d'évaluation
- Liquidación Salarios en Colombia.Document40 pagesLiquidación Salarios en Colombia.Jhonnier CarreraPas encore d'évaluation
- Vol I Cap 2 Soc Orig CaribeDocument24 pagesVol I Cap 2 Soc Orig CaribeHernán Reales Vega100% (1)
- El ferrocarril de Bolívar y la consolidación del puerto de BarranquillaDocument26 pagesEl ferrocarril de Bolívar y la consolidación del puerto de BarranquillaHernán Reales VegaPas encore d'évaluation
- Ensayo Leslie Bethell Capitulo 3 - LatinoamericaDocument2 pagesEnsayo Leslie Bethell Capitulo 3 - LatinoamericaHernán Reales Vega0% (1)
- BorrachoDocument1 pageBorrachoHernán Reales VegaPas encore d'évaluation
- Apoptosis NecrosisDocument6 pagesApoptosis NecrosisajeinsonPas encore d'évaluation
- ApoptosisDocument9 pagesApoptosisHernán Reales VegaPas encore d'évaluation
- Salud mental y adolescenciaDocument23 pagesSalud mental y adolescenciaKevin Gonzalo Leon L.Pas encore d'évaluation
- Diagnostico SituacionalDocument28 pagesDiagnostico SituacionalCarolina Vasquez MirandaPas encore d'évaluation
- Unidad 13 - Nivel IvDocument7 pagesUnidad 13 - Nivel IvOscar Valdes FallaPas encore d'évaluation
- Prevencion de Riesgos Laborales en La ConstrucciónDocument357 pagesPrevencion de Riesgos Laborales en La ConstrucciónomarblozadaPas encore d'évaluation
- Aumento trastornos mentales ColombiaDocument3 pagesAumento trastornos mentales ColombiaTatiana MoralesPas encore d'évaluation
- Guía de Atención en Salud Mental en Emergencias y DesastresDocument141 pagesGuía de Atención en Salud Mental en Emergencias y DesastresJuan Santiago BotelloPas encore d'évaluation
- Manual de Seguridad EscolarDocument19 pagesManual de Seguridad EscolarAli OrozcoPas encore d'évaluation
- Enfermería Del Adulto y Anciano IIDocument4 pagesEnfermería Del Adulto y Anciano IIStefano PastoriniPas encore d'évaluation
- Factores psicosociales y salud mental en enfermería quirúrgicaDocument112 pagesFactores psicosociales y salud mental en enfermería quirúrgicaMelissa OchoaPas encore d'évaluation
- Recuerdos de la infanciaDocument140 pagesRecuerdos de la infanciaVeru Dugarte0% (1)
- 20 Claudia Elena Morazán SilvaDocument27 pages20 Claudia Elena Morazán SilvaClaudia Elena Morazán SilvaPas encore d'évaluation
- Trabajo Grupo8Document15 pagesTrabajo Grupo8Fernando Siancas ShimokawaPas encore d'évaluation
- Línea Del Tiempo Avances MédicosDocument11 pagesLínea Del Tiempo Avances MédicosPaulinaPas encore d'évaluation
- Evaluación y tratamiento de las disfunciones sexuales femeninasDocument140 pagesEvaluación y tratamiento de las disfunciones sexuales femeninasSebax CarvaramiPas encore d'évaluation
- Relaciones HumanasDocument42 pagesRelaciones Humanasandrea do santosPas encore d'évaluation
- Fase1 - Julieth Ramos - 403023 - 479Document7 pagesFase1 - Julieth Ramos - 403023 - 479Yulirth RamosPas encore d'évaluation
- Guia en Materia de Riesgos Laborales Del Trabajo en Plataformas Digitales 2020Document51 pagesGuia en Materia de Riesgos Laborales Del Trabajo en Plataformas Digitales 2020Xabier Dominguez Garcia de AndoinPas encore d'évaluation
- En Una Entrevista Psicológica Se Produce Una Relación Entre Dos o Más PersonasDocument17 pagesEn Una Entrevista Psicológica Se Produce Una Relación Entre Dos o Más PersonasHoselyta BravoPas encore d'évaluation
- Memoria Autobiográfica, Trauma y Construcción Del YoDocument161 pagesMemoria Autobiográfica, Trauma y Construcción Del YoJuan Carlos PaniaguaPas encore d'évaluation
- Betty NeumanDocument14 pagesBetty NeumanTiendita UnicaPas encore d'évaluation
- Técnicas para Controlar El EstresDocument1 pageTécnicas para Controlar El EstresFranciscaPas encore d'évaluation
- Teorías explicativas del maltrato a la mujerDocument24 pagesTeorías explicativas del maltrato a la mujerArismendy Rodriguez100% (1)
- Papel de La Resiliencia en Personas Mayores InstitucionalizadasDocument32 pagesPapel de La Resiliencia en Personas Mayores InstitucionalizadasJonathan RGPas encore d'évaluation