Vous aimerez peut-être aussi
- Sistema de EcuacionesDocument11 pagesSistema de EcuacionesPatricio Fernando Cornejo CastroPas encore d'évaluation
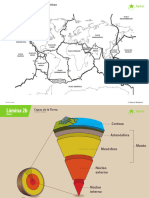
- 2022 7 basico Ciencias Naturales Modulo 3 Clase 2 LaminasDocument3 pages2022 7 basico Ciencias Naturales Modulo 3 Clase 2 LaminasPatricio Fernando Cornejo CastroPas encore d'évaluation
- AnaliDocument35 pagesAnaliPatricio Fernando Cornejo CastroPas encore d'évaluation
- Disertacion Sobre El MundialDocument1 pageDisertacion Sobre El MundialPatricio Fernando Cornejo CastroPas encore d'évaluation
- PRT-711.02-011 V1 Determinación de Cenizas TotalesDocument2 pagesPRT-711.02-011 V1 Determinación de Cenizas TotalesPatricio Fernando Cornejo CastroPas encore d'évaluation
- Tutor RMNDocument59 pagesTutor RMNfabiolin9Pas encore d'évaluation
- Edafo Tipeo 2 Prueba 3Document14 pagesEdafo Tipeo 2 Prueba 3Patricio Fernando Cornejo CastroPas encore d'évaluation
- Ej Poleas, Engranajes y PotenciasDocument4 pagesEj Poleas, Engranajes y PotenciasPatricio Fernando Cornejo CastroPas encore d'évaluation
- Cap8 Zegarra Numeros ComplejosDocument30 pagesCap8 Zegarra Numeros ComplejosAlexis ValladaresPas encore d'évaluation
- Capítulo XIVDocument3 pagesCapítulo XIVPatricio Fernando Cornejo CastroPas encore d'évaluation
- Erikson OchoedadesDocument27 pagesErikson OchoedadesPatricio Fernando Cornejo CastroPas encore d'évaluation
- Apuntes MagdalenoDocument14 pagesApuntes MagdalenoJorge Fernando Vega LugoPas encore d'évaluation
- Apunte Tecno Minera Oto o 2019Document189 pagesApunte Tecno Minera Oto o 2019jose peñaPas encore d'évaluation
- Comportamiento extremo del fuegoDocument28 pagesComportamiento extremo del fuegoFabian IrarrazabalPas encore d'évaluation
- Protocolo para La Determinacion de Hongos Micorrizicos ArbuscularesDocument51 pagesProtocolo para La Determinacion de Hongos Micorrizicos ArbuscularesCristhiam Bermudez MatusPas encore d'évaluation
- Distribucion - Yacimientos - Sonora Ochoa Etal 2009Document6 pagesDistribucion - Yacimientos - Sonora Ochoa Etal 2009Diamond100% (2)
- Radiación solar: proceso de balance y distribuciónDocument6 pagesRadiación solar: proceso de balance y distribuciónEdgar Agustin Ortellado RamírezPas encore d'évaluation
- Cuestionario BotanicaDocument6 pagesCuestionario BotanicaEmmanuel GutiérrezPas encore d'évaluation
- Eissen Et AlDocument3 pagesEissen Et AlPaúl CabezasPas encore d'évaluation
- Estudio impacto ambiental frakingDocument4 pagesEstudio impacto ambiental frakinglorena lopezPas encore d'évaluation
- Cuestionario GeotecniaDocument28 pagesCuestionario Geotecniaandres torresPas encore d'évaluation
- Planeación Movimiento OscilatorioDocument12 pagesPlaneación Movimiento OscilatorioJulian VargasPas encore d'évaluation
- Conclusiones Microzonificacion SismicaDocument34 pagesConclusiones Microzonificacion SismicagilmarPas encore d'évaluation
- Charla Uso y Manejo de ExtintoresDocument1 pageCharla Uso y Manejo de Extintoresperla castilloPas encore d'évaluation
- Morín - Astrología Galica, libro 18Document49 pagesMorín - Astrología Galica, libro 18Fernando Luis OrtizPas encore d'évaluation
- La Tabla Periodica Y Las Sustancias Químicas: Ing. Gerardo Flores RujelDocument34 pagesLa Tabla Periodica Y Las Sustancias Químicas: Ing. Gerardo Flores RujelJohao Olaya espinozaPas encore d'évaluation
- Proyecto de QuimicaDocument6 pagesProyecto de QuimicaGENOCIDALPas encore d'évaluation
- FISICA Maestra Nastya Jesus DamianDocument1 pageFISICA Maestra Nastya Jesus DamianLuis GutierrezPas encore d'évaluation
- Ejercicios FfyqqDocument14 pagesEjercicios FfyqqAlejandro Marín Sánchez50% (2)
- QUE ES FENOMENO DEL El - Niño - Febrero - 2019Document6 pagesQUE ES FENOMENO DEL El - Niño - Febrero - 2019Pedro BacaPas encore d'évaluation
- Metereorizacion y Sus TiposDocument7 pagesMetereorizacion y Sus TiposMillie SandovalPas encore d'évaluation
- Experimento 2Document25 pagesExperimento 2AlvaroGabrielRivasDíazPas encore d'évaluation
- Clase 3 - Avaluo de Cargas - EtabsDocument33 pagesClase 3 - Avaluo de Cargas - EtabsJorge Alexander ARGUELLEZPas encore d'évaluation
- Lutitas PDFDocument6 pagesLutitas PDFrenatoPas encore d'évaluation
- Relacion de Mineros Formales Modif Actualizado.Document1 pageRelacion de Mineros Formales Modif Actualizado.Zarai LesliePas encore d'évaluation
- Evaluación de Características de La MateriaDocument2 pagesEvaluación de Características de La MateriaMartha Melida Piedrahta Llano0% (1)
- Aguas SubterraneasDocument2 pagesAguas Subterraneasmichael camiloPas encore d'évaluation
- Actividad 3Document2 pagesActividad 3jhailer poloPas encore d'évaluation
- Potencial ZDocument15 pagesPotencial ZUriel Mqz100% (1)
- Serrano CPFDocument162 pagesSerrano CPFEliane Melany Prado RivasPas encore d'évaluation
- Sobreexplotación bosques Michoacán cultivo aguacateDocument3 pagesSobreexplotación bosques Michoacán cultivo aguacateandresPas encore d'évaluation