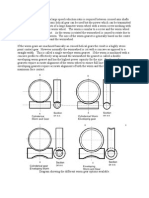
Vous aimerez peut-être aussi
- SX001a en EU Example Simply Supported Laterally UnrestrainedDocument9 pagesSX001a en EU Example Simply Supported Laterally UnrestrainedJamesPas encore d'évaluation
- Fracture Toughness Testing - TwiDocument5 pagesFracture Toughness Testing - TwiSergio MunhosPas encore d'évaluation
- Fundamentals of Electromagnetic Theory, Khunita, PHIDocument11 pagesFundamentals of Electromagnetic Theory, Khunita, PHIkofir100% (1)
- A Worm GearDocument19 pagesA Worm GearPetrus Malailak0% (1)
- Flange LeakageDocument48 pagesFlange LeakageAnjani Prabhakar83% (6)
- Experiment 4: Work, Power and Energy Laboratory Report Group 5Document6 pagesExperiment 4: Work, Power and Energy Laboratory Report Group 5Socorro DuquePas encore d'évaluation
- The Chemistry of Natural Products: 6: Plenary Lectures Presented at the Sixth International Symposium on the Chemistry of Natural ProductsD'EverandThe Chemistry of Natural Products: 6: Plenary Lectures Presented at the Sixth International Symposium on the Chemistry of Natural ProductsPas encore d'évaluation
- 5 Domon 1984Document6 pages5 Domon 1984Enciu MariaPas encore d'évaluation
- Ajc 25 5 47Document4 pagesAjc 25 5 47thiên vũ hoàngPas encore d'évaluation
- $yano 3Document3 pages$yano 3ashokPas encore d'évaluation
- Drugs of The Future 2002, 27 (2) 143-158Document16 pagesDrugs of The Future 2002, 27 (2) 143-158Rajesh TammanaPas encore d'évaluation
- No Card IcinDocument12 pagesNo Card IcinHazrati UmmiPas encore d'évaluation
- Hoshino 1996Document7 pagesHoshino 1996ivanjavierlozadayala178Pas encore d'évaluation
- Novel Oxidation Products of Anthocyanins in Black RiceDocument6 pagesNovel Oxidation Products of Anthocyanins in Black RiceMasjid Nurul ImanPas encore d'évaluation
- Azo CompsDocument5 pagesAzo CompsarooojfataimaPas encore d'évaluation
- Mode of ChiralityDocument8 pagesMode of ChiralitychemistPas encore d'évaluation
- A New Ent-Clerodane Diterpene From The Earial Parts of Baccharis Gaudichaudiana (2003)Document3 pagesA New Ent-Clerodane Diterpene From The Earial Parts of Baccharis Gaudichaudiana (2003)TàiNguyễnThànhPas encore d'évaluation
- CoumarinsDocument5 pagesCoumarinsAmr El DemerdashPas encore d'évaluation
- Tanacetumpseudoachillea: 10011. N o MechanicalDocument2 pagesTanacetumpseudoachillea: 10011. N o MechanicalLiendo Polanco GustavoPas encore d'évaluation
- Total Synthesis of 7-'4C-(i)-ColchicineDocument11 pagesTotal Synthesis of 7-'4C-(i)-ColchicinePaula LomboPas encore d'évaluation
- Synthesis and Some Transformations of (-) - CarveolDocument6 pagesSynthesis and Some Transformations of (-) - CarveolEman MifsudPas encore d'évaluation
- BBTK1Document4 pagesBBTK1HàPas encore d'évaluation
- Xanthones and Flavonoids from Polygala caudata RootsDocument3 pagesXanthones and Flavonoids from Polygala caudata RootsTuấn Nguyen AnhPas encore d'évaluation
- JOC 1978 (43) 2320 - SasakiDocument6 pagesJOC 1978 (43) 2320 - SasakigioLXVPas encore d'évaluation
- Rhombifoline and 5,6-Dehydrolupanine From: Anagyrus Foetida LDocument5 pagesRhombifoline and 5,6-Dehydrolupanine From: Anagyrus Foetida Lchirag sabhayaPas encore d'évaluation
- Articulo 4Document5 pagesArticulo 4Viviana TorresPas encore d'évaluation
- $RII5WWODocument3 pages$RII5WWOjiracioPas encore d'évaluation
- Aromatic Compounds From Delphinium Venulosum: in Reoisedform 29 May 1991)Document2 pagesAromatic Compounds From Delphinium Venulosum: in Reoisedform 29 May 1991)Dr-Muhammad Imran TousifPas encore d'évaluation
- Nascimento2003 PDFDocument5 pagesNascimento2003 PDFMita KurniatiPas encore d'évaluation
- Penicillium Citrinum F5: 2,3,4-Trimethyl-5,7-Dihydroxy-2,3-Dihydrobenzofuran, A Novel Antioxidant, FromDocument2 pagesPenicillium Citrinum F5: 2,3,4-Trimethyl-5,7-Dihydroxy-2,3-Dihydrobenzofuran, A Novel Antioxidant, FromShiraz ButtPas encore d'évaluation
- Antiviral Diterpenes From Salvia OfficinalisDocument3 pagesAntiviral Diterpenes From Salvia OfficinalisGuaguanconPas encore d'évaluation
- Art:10 1007/BF00846627Document3 pagesArt:10 1007/BF00846627Tabitha MoorePas encore d'évaluation
- Oxido-And Dioxidovanadium V Complexes Wi PDFDocument10 pagesOxido-And Dioxidovanadium V Complexes Wi PDFAdnan Ahmed ChahalPas encore d'évaluation
- Highly Selective Oxalate - Membrane Electrode Based On (Cul) (Ac)Document10 pagesHighly Selective Oxalate - Membrane Electrode Based On (Cul) (Ac)Hani KhuludPas encore d'évaluation
- Synthesis and Characterization of New Pyrazoline and Isoxazoline Derivatives Based On FluoreneDocument7 pagesSynthesis and Characterization of New Pyrazoline and Isoxazoline Derivatives Based On FluorenevivimeinaPas encore d'évaluation
- Molecules 02 00003Document4 pagesMolecules 02 00003Félix Álvarez de BrogliePas encore d'évaluation
- 1 s2.0 S0020169305006274 Main PDFDocument5 pages1 s2.0 S0020169305006274 Main PDFhenry martinez quiñonezPas encore d'évaluation
- JNatProd66 (2003) 1101Document3 pagesJNatProd66 (2003) 1101Ana Paula SantosPas encore d'évaluation
- 1 s2.0 S0020169305004287 Main PDFDocument9 pages1 s2.0 S0020169305004287 Main PDFhenry martinez quiñonezPas encore d'évaluation
- Krukovine, A New Bisbenzylisoquinoline Alkaloid From Abuta SplendidaDocument3 pagesKrukovine, A New Bisbenzylisoquinoline Alkaloid From Abuta SplendidaDavid ScoPas encore d'évaluation
- Synthesis of 2-Amino-3-Ethoxycarbonylthiophene DerivativesDocument3 pagesSynthesis of 2-Amino-3-Ethoxycarbonylthiophene Derivativesborons234Pas encore d'évaluation
- Cichorium intybus constituents and activitiesDocument6 pagesCichorium intybus constituents and activitiesreza rezaiePas encore d'évaluation
- Aspergillus Fumigatus: Novel Reduction and Hydroxylation Products Formed by From Reichstein's Substance SDocument5 pagesAspergillus Fumigatus: Novel Reduction and Hydroxylation Products Formed by From Reichstein's Substance ShelmetuserPas encore d'évaluation
- Tugas TIDocument5 pagesTugas TIMoch Alie MuchitPas encore d'évaluation
- Erna Fitriana AlfantiDocument6 pagesErna Fitriana AlfantiIzam M. FalahPas encore d'évaluation
- Amodiaquine HydrochlorideDocument31 pagesAmodiaquine HydrochlorideJulia TaranchukPas encore d'évaluation
- Djokic JCS Perkin Trans.1 1986 Pp1881Document10 pagesDjokic JCS Perkin Trans.1 1986 Pp1881Mario Micciarelli100% (1)
- 6-Methoxybenzoxazolinone and Triterpenoids from Roots of Scoparia dulcisDocument3 pages6-Methoxybenzoxazolinone and Triterpenoids from Roots of Scoparia dulcisMinyty LePas encore d'évaluation
- Stereospecific Total Synthesis of 9-Aminoanthracyclines: (+)-9-Amino-9-deoxydaunomycin and Related CompoundsDocument9 pagesStereospecific Total Synthesis of 9-Aminoanthracyclines: (+)-9-Amino-9-deoxydaunomycin and Related CompoundsDr. P. Ramu SridharPas encore d'évaluation
- Synthesis, Spectral Characterization and In-Vitro Analysis of Some Novel Title Chalcones DerivativesDocument5 pagesSynthesis, Spectral Characterization and In-Vitro Analysis of Some Novel Title Chalcones DerivativesSudhanshu Kumar JhaPas encore d'évaluation
- Cydonia Vulg ArticleDocument6 pagesCydonia Vulg Articlebm rdPas encore d'évaluation
- Zhao J DKK 2001 - Three New Triterpene Saponin From The Seeds of A, ChinensisDocument3 pagesZhao J DKK 2001 - Three New Triterpene Saponin From The Seeds of A, ChinensisSeptynelya ThenuPas encore d'évaluation
- Struktur AcetogeninDocument7 pagesStruktur Acetogeninpepe_onetPas encore d'évaluation
- A Practical Synthesis of (-) - Kainic AcidDocument11 pagesA Practical Synthesis of (-) - Kainic AcidNikhil SarpatePas encore d'évaluation
- New method for the synthesis of macrocyclic compounds. Communication 3. Intramolecular alkylation of 2- (ethoxycarbonylacetyl) -5- (ω-iodoalkyl) thiophenesDocument7 pagesNew method for the synthesis of macrocyclic compounds. Communication 3. Intramolecular alkylation of 2- (ethoxycarbonylacetyl) -5- (ω-iodoalkyl) thiophenesSiwar AitPas encore d'évaluation
- Limonoids From Cedrela SinensisDocument7 pagesLimonoids From Cedrela Sinensisnishi@sainiPas encore d'évaluation
- NPC Natural Product Communications: Phenolic Constituents From Xyloselinum LeonidiiDocument2 pagesNPC Natural Product Communications: Phenolic Constituents From Xyloselinum LeonidiiDiệp Vĩnh TânPas encore d'évaluation
- Five New Iboga Alkaloids From Tabernaemontana Corymbosa: Toh-Seok Kam and Kooi-Mow SimDocument4 pagesFive New Iboga Alkaloids From Tabernaemontana Corymbosa: Toh-Seok Kam and Kooi-Mow SimRichard PortillooPas encore d'évaluation
- Deve HatDocument5 pagesDeve HatOkky Winang SaktyawanPas encore d'évaluation
- Dinoflagellate Luciferin Is Structurally Related To ChlorophyllDocument4 pagesDinoflagellate Luciferin Is Structurally Related To ChlorophylljavierPas encore d'évaluation
- Synthesis of The Algicide BacillamideDocument7 pagesSynthesis of The Algicide Bacillamiderajesh kothariPas encore d'évaluation
- A Chemoenzymatic Total Synthesis of The Structure Assigned To The Alkaloid (+) - MontabuphineDocument41 pagesA Chemoenzymatic Total Synthesis of The Structure Assigned To The Alkaloid (+) - MontabuphineLê MinhPas encore d'évaluation
- Iridoid and Phenylethanoid Glycosides From Euphrasia Pectinata (#142919) - 124342Document10 pagesIridoid and Phenylethanoid Glycosides From Euphrasia Pectinata (#142919) - 124342Hashemi Akhter100% (1)
- R.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeDocument5 pagesR.A. Heacock and M.E. Mahon - The Chemistry of The "Aminochromes" Part II: The Preparation, Paper Chromatography, and Spectroscopic Properties of Pure Adrenolutin The Infrared Spectrum of AdrenochromeGummyColaPas encore d'évaluation
- Supporting Information ForDocument28 pagesSupporting Information ForMAnugrahRizkyPPas encore d'évaluation
- Synthesis, Antibacterial and Antifungal Evlaution of Novel Pyrazoline DerivativesDocument6 pagesSynthesis, Antibacterial and Antifungal Evlaution of Novel Pyrazoline DerivativesNexi anessaPas encore d'évaluation
- Attenuation in Optical FiberDocument22 pagesAttenuation in Optical Fibermanishsoni30Pas encore d'évaluation
- PHYS 101 / Conservation of Linear Momentum Experiment 5 Data SheetDocument5 pagesPHYS 101 / Conservation of Linear Momentum Experiment 5 Data SheetAlpPas encore d'évaluation
- Classical Theory of Rayleigh and Raman Scattering 18pDocument18 pagesClassical Theory of Rayleigh and Raman Scattering 18psatishmaanPas encore d'évaluation
- Resource Letter - Van Der Waals and Casimir-Polder Forces PDFDocument27 pagesResource Letter - Van Der Waals and Casimir-Polder Forces PDFthelordxorPas encore d'évaluation
- Chapter 8Document9 pagesChapter 8siddpawar08Pas encore d'évaluation
- SW#2 Integrals of Trigonometric FunctionsDocument1 pageSW#2 Integrals of Trigonometric FunctionsRachel MonesPas encore d'évaluation
- Albert EinsteinDocument1 pageAlbert EinsteinAnggi100% (1)
- Magnetic Characteristics of Different Mukhi Rudraksha Beads: A Comparative AnalysisDocument6 pagesMagnetic Characteristics of Different Mukhi Rudraksha Beads: A Comparative AnalysisMariappan RPas encore d'évaluation
- The LengthDocument1 pageThe LengthRAIZZ67% (3)
- CE-515: Design of Steel StructuresDocument15 pagesCE-515: Design of Steel StructuresZiaullahPas encore d'évaluation
- Iit Chemistry 4Document11 pagesIit Chemistry 4Shailendra Agarwal100% (1)
- Namma Kalvi 12th Physics Unit 1 Sura Guide em 214945Document62 pagesNamma Kalvi 12th Physics Unit 1 Sura Guide em 214945Lakshitha SivakumarPas encore d'évaluation
- Unsolved Problems in ScienceDocument2 pagesUnsolved Problems in ScienceJ GPas encore d'évaluation
- Virtual MethodDocument1 pageVirtual Methodsuganthi1711Pas encore d'évaluation
- Magnetic High Tension SeparatorsDocument37 pagesMagnetic High Tension SeparatorsAdel Niño Liza IliganPas encore d'évaluation
- Questions & Answers On Rating and Loss DissipationDocument25 pagesQuestions & Answers On Rating and Loss Dissipationkibrom atsbhaPas encore d'évaluation
- 1408100045-Nila Huda-Thesaurus of XRDDocument3 pages1408100045-Nila Huda-Thesaurus of XRDNilaHudaBaqirPas encore d'évaluation
- Casting RefDocument20 pagesCasting RefNavdeep GillPas encore d'évaluation
- Chapter 4 Fourier Optics: D D y X J F y X F FDocument14 pagesChapter 4 Fourier Optics: D D y X J F y X F FansarixxxPas encore d'évaluation
- Growth Kinetics and Mechanical Properties of Boride Laye 2013 Surface and CoDocument13 pagesGrowth Kinetics and Mechanical Properties of Boride Laye 2013 Surface and CoFRANCISCO TORRESPas encore d'évaluation
- Replacement of Coarse Aggregate by Cinder Aggregate in Light Weight Concrete.Document27 pagesReplacement of Coarse Aggregate by Cinder Aggregate in Light Weight Concrete.Parashu Ram Neo100% (1)
- Great Formulas Explained Physics Mathematics EconomicsDocument135 pagesGreat Formulas Explained Physics Mathematics EconomicsBrody ChanPas encore d'évaluation
- Astm E748 2016Document11 pagesAstm E748 2016Manoj VishwakarmaPas encore d'évaluation