Vous aimerez peut-être aussi
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Babylonian Sacred Words of Power - Carl Nagel 1988Document66 pagesBabylonian Sacred Words of Power - Carl Nagel 1988leplafonfagnon90% (99)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5795)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Model Village PDFDocument6 pagesModel Village PDFakshat100% (1)
- ESG Assignment Part 2: Shareholder ActivismDocument6 pagesESG Assignment Part 2: Shareholder ActivismParesh ShrivastavaPas encore d'évaluation
- Capillary PressureDocument12 pagesCapillary PressureamahaminerPas encore d'évaluation
- Molecular DockingDocument13 pagesMolecular DockingAnand MauryaPas encore d'évaluation
- Spinning CalculationDocument178 pagesSpinning Calculationamboklate69% (16)
- HISTORY AND CURRENT STATUS OF CHEMOINFORMATICSsDocument10 pagesHISTORY AND CURRENT STATUS OF CHEMOINFORMATICSsAnand MauryaPas encore d'évaluation
- Factorial ExperimentsDocument16 pagesFactorial ExperimentsAnand MauryaPas encore d'évaluation
- cDNA MicrorrayDocument13 pagescDNA MicrorrayAnand MauryaPas encore d'évaluation
- DeputationDocument8 pagesDeputationAnand MauryaPas encore d'évaluation
- Plant Physiol. 2001 A 160 3Document4 pagesPlant Physiol. 2001 A 160 3Anand MauryaPas encore d'évaluation
- Medical Database SystemDocument2 pagesMedical Database SystemAnand MauryaPas encore d'évaluation
- Answers Answers Answers Answers Answers: T Est - 1Document9 pagesAnswers Answers Answers Answers Answers: T Est - 1Anand MauryaPas encore d'évaluation
- B.tech - Non Credit Courses For 2nd Year StudentsDocument4 pagesB.tech - Non Credit Courses For 2nd Year StudentsNishant MishraPas encore d'évaluation
- Chapter 12 My Nursing Test BanksDocument10 pagesChapter 12 My Nursing Test Banksنمر نصار100% (1)
- INV201 ReadParameterDocument3 pagesINV201 ReadParameterRaul ArbaniesPas encore d'évaluation
- Affective DomainDocument3 pagesAffective DomainJm Enriquez Dela Cruz50% (2)
- Solarbotics Wheel Watcher Encoder ManualDocument10 pagesSolarbotics Wheel Watcher Encoder ManualYash SharmaPas encore d'évaluation
- Rehabilitation Programs On The Behavior of Juveniles in Manga Children's Remand Home, Nyamira County - KenyaDocument7 pagesRehabilitation Programs On The Behavior of Juveniles in Manga Children's Remand Home, Nyamira County - KenyaInternational Journal of Innovative Science and Research TechnologyPas encore d'évaluation
- Observation Method in Qualitative ResearchDocument42 pagesObservation Method in Qualitative Researchsantoshipoudel08Pas encore d'évaluation
- Đề Thi Tham Khảo (Đề thi có 5 trang) : 6: The flood victims 7: Many 8Document7 pagesĐề Thi Tham Khảo (Đề thi có 5 trang) : 6: The flood victims 7: Many 8Quang Đặng HồngPas encore d'évaluation
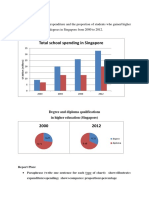
- Total School Spending in Singapore: Task 1 020219Document6 pagesTotal School Spending in Singapore: Task 1 020219viper_4554Pas encore d'évaluation
- Types of Dance Steps and Positions PDFDocument11 pagesTypes of Dance Steps and Positions PDFRather NotPas encore d'évaluation
- Tips Experiments With MatlabDocument190 pagesTips Experiments With MatlabVishalPas encore d'évaluation
- 1300 Rev01Document15 pages1300 Rev01Manuel AltamiranoPas encore d'évaluation
- Money, Sex, and Power: Toward A Feminist Historical MaterialismDocument2 pagesMoney, Sex, and Power: Toward A Feminist Historical MaterialismddwererPas encore d'évaluation
- Practical Means For Energy-Based Analyses of Disproportionate Collapse PotentialDocument13 pagesPractical Means For Energy-Based Analyses of Disproportionate Collapse PotentialNhut NguyenPas encore d'évaluation
- Oracle Database Administrator TasksDocument2 pagesOracle Database Administrator TasksPratik GandhiPas encore d'évaluation
- Listening Test 1.1Document2 pagesListening Test 1.1momobear152Pas encore d'évaluation
- Manufacturing Technology - Metrology: Dr.B.Ramamoorthy Professor Manufacturing Engg. Section Iitmadras 600 036Document22 pagesManufacturing Technology - Metrology: Dr.B.Ramamoorthy Professor Manufacturing Engg. Section Iitmadras 600 036Ramasubramanian KannanPas encore d'évaluation
- 27 EtdsDocument29 pages27 EtdsSuhag PatelPas encore d'évaluation
- Problem Solving and Algorithms: Problems, Solutions, and ToolsDocument12 pagesProblem Solving and Algorithms: Problems, Solutions, and ToolsskarthikpriyaPas encore d'évaluation
- ESM Module1 GE3 TCWDocument12 pagesESM Module1 GE3 TCWDeserie Aguilar DugangPas encore d'évaluation
- Lesson 2 (Locating Main Ideas)Document18 pagesLesson 2 (Locating Main Ideas)Glyneth Dela TorrePas encore d'évaluation
- Global 1 Syllabus Fall 2014Document5 pagesGlobal 1 Syllabus Fall 2014Henry WangPas encore d'évaluation
- Momi BhattacharyaDocument59 pagesMomi BhattacharyamohchinbokhtiyarPas encore d'évaluation
- The Structure of Deception: Validation of The Lying Profile QuestionnaireDocument16 pagesThe Structure of Deception: Validation of The Lying Profile QuestionnaireNancy DrewPas encore d'évaluation
- Comparision BRC IFS QMS 22K From BV PDFDocument44 pagesComparision BRC IFS QMS 22K From BV PDFAhmedElSayedPas encore d'évaluation