Vous aimerez peut-être aussi
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5795)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- TCSP10403R0Document30 pagesTCSP10403R0BADRI VENKATESHPas encore d'évaluation
- Electrical Properties of Lipid Bilayers-Veronica PRISECARIUDocument12 pagesElectrical Properties of Lipid Bilayers-Veronica PRISECARIUVeronica PrisecariuPas encore d'évaluation
- 2 2) Cos 1 (2 2) Cos 1 (: L6 - Surface Tension of BiomaterialsDocument2 pages2 2) Cos 1 (2 2) Cos 1 (: L6 - Surface Tension of BiomaterialsVeronica PrisecariuPas encore d'évaluation
- L4 - FTIR Spectroscopy Study of Protein DenaturationDocument4 pagesL4 - FTIR Spectroscopy Study of Protein DenaturationVeronica PrisecariuPas encore d'évaluation
- L2 - Surface Roughness of Medical Implants: Analysis of AFM ImagesDocument4 pagesL2 - Surface Roughness of Medical Implants: Analysis of AFM ImagesVeronica PrisecariuPas encore d'évaluation
- Building Applied Natural Language GenerationDocument32 pagesBuilding Applied Natural Language Generationrat86Pas encore d'évaluation
- Nitrile RubberDocument1 pageNitrile RubberMohamedPas encore d'évaluation
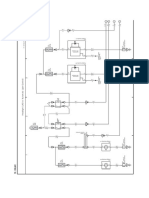
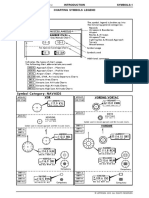
- Overall EWD Vehicle Exterior Rear Fog LightDocument10 pagesOverall EWD Vehicle Exterior Rear Fog Lightgabrielzinho43Pas encore d'évaluation
- VP422 HDTV10A Service Manual PDFDocument25 pagesVP422 HDTV10A Service Manual PDFDan Prewitt0% (1)
- CVision AVR Man3 PDFDocument513 pagesCVision AVR Man3 PDFsigiloPas encore d'évaluation
- UL FM Vertical Indicator Post Maintenance and Operation Manual - Fire Protection - Models 5400 & 5450Document7 pagesUL FM Vertical Indicator Post Maintenance and Operation Manual - Fire Protection - Models 5400 & 5450Juan Jose Teodoro AguilarPas encore d'évaluation
- Lesson 3 - Equipment and FacilitiesDocument15 pagesLesson 3 - Equipment and FacilitiesRishiel Dimple BalonesPas encore d'évaluation
- Jeppesen Charts LegendsDocument34 pagesJeppesen Charts LegendsFatih OguzPas encore d'évaluation
- TR 541 2Document78 pagesTR 541 2Omar Marghani SalmaPas encore d'évaluation
- High Voltage Engineering MCQsDocument6 pagesHigh Voltage Engineering MCQsSubrahmanyam Adda50% (2)
- Cooling Coil SizingDocument4 pagesCooling Coil SizingRanu JanuarPas encore d'évaluation
- Gad Ad2 J10k1020c27a 200a201Document1 pageGad Ad2 J10k1020c27a 200a201AbdulPas encore d'évaluation
- GERMAN Embassy ContractDocument79 pagesGERMAN Embassy ContractVE.03 QELPas encore d'évaluation
- Understanding The 808Document3 pagesUnderstanding The 808Israel LuRuvalPas encore d'évaluation
- Is 13935 2009 PDFDocument33 pagesIs 13935 2009 PDFManojKumawatRjPas encore d'évaluation
- Tunnels and Suport SystemsDocument33 pagesTunnels and Suport SystemsShella Marie Nartatez-NiroPas encore d'évaluation
- Reverb DesignDocument9 pagesReverb DesignSerj PoltavskiPas encore d'évaluation
- Penawaran Fility-70 Lebar 9 - ArmayaDocument2 pagesPenawaran Fility-70 Lebar 9 - ArmayaSketchUp panduanPas encore d'évaluation
- How To Make Acrylic Bending Machine DIY PVC Sheet Bender Mistry MakeToolDocument8 pagesHow To Make Acrylic Bending Machine DIY PVC Sheet Bender Mistry MakeToolnardusPas encore d'évaluation
- Taglio Sez Circ 2Document3 pagesTaglio Sez Circ 2mariorossi_4Pas encore d'évaluation
- The Radio ClubDocument7 pagesThe Radio ClubDom CasualPas encore d'évaluation
- Mine Survey CertificationDocument37 pagesMine Survey CertificationAgustin Eliasta Ginting100% (1)
- Cable Ties CatalogDocument60 pagesCable Ties CatalogRvPas encore d'évaluation
- Mycom TecnicaDocument21 pagesMycom TecnicaTeuku Mukhriza100% (1)
- PolyIT AdjustmentDocument10 pagesPolyIT AdjustmentSwami MeeraPas encore d'évaluation
- Irgb 4064 DPBFDocument11 pagesIrgb 4064 DPBFKrista TranPas encore d'évaluation
- VCD-D ManualDocument13 pagesVCD-D ManualnimmuhkPas encore d'évaluation
- Crystal StructuresDocument54 pagesCrystal StructuresyashvantPas encore d'évaluation
- R05411101 ImageprocessingandpatternrecognitionDocument4 pagesR05411101 ImageprocessingandpatternrecognitionSamiullah MohammedPas encore d'évaluation