Vous aimerez peut-être aussi
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Chapter 41 - Thoracic Outlet Syndrome SynonymsDocument8 pagesChapter 41 - Thoracic Outlet Syndrome SynonymsPiero Massafra100% (1)
- MEDICAL SURGICAL NURSING REVIEWDocument7 pagesMEDICAL SURGICAL NURSING REVIEWeloisa mae gementizaPas encore d'évaluation
- BookDocument211 pagesBookAngela KocevskaPas encore d'évaluation
- CNS: Anatomy, Cases of Trauma, Congenital, Infection, Degeneration & TumorsDocument29 pagesCNS: Anatomy, Cases of Trauma, Congenital, Infection, Degeneration & Tumorsamir hamzahPas encore d'évaluation
- Biological Level of Analysis Research GuideDocument45 pagesBiological Level of Analysis Research GuidePhiline Everts100% (2)
- Irritable Bowel Syndrome Natural SolutionsDocument25 pagesIrritable Bowel Syndrome Natural SolutionsBrettPas encore d'évaluation
- Full PDFDocument399 pagesFull PDFTeresa Marie Yap CorderoPas encore d'évaluation
- Head Toe Physical AssessmentDocument2 pagesHead Toe Physical Assessmentzbestgurl100% (2)
- Microsoft Word - Endodontic - MishapsDocument20 pagesMicrosoft Word - Endodontic - MishapsShufeiPas encore d'évaluation
- Jump Height Loss As An Indicator of Fatigue During Sprint TrainingDocument11 pagesJump Height Loss As An Indicator of Fatigue During Sprint TrainingLevyPas encore d'évaluation
- Part of Brain 2Document2 pagesPart of Brain 2PonCut Teuku AchyarPas encore d'évaluation
- MIDTERM Pilar College Anatomy Physiology Final EditDocument41 pagesMIDTERM Pilar College Anatomy Physiology Final EditYeona BaePas encore d'évaluation
- Human Motivation Edited2Document23 pagesHuman Motivation Edited2jingoPas encore d'évaluation
- Kidney PowerPoint Presentation TemplateDocument14 pagesKidney PowerPoint Presentation TemplateMedicinePPT100% (1)
- OADocument27 pagesOADarkKnighthere100% (1)
- 08 DN 005 BA Int OkDocument64 pages08 DN 005 BA Int OkvbogachevPas encore d'évaluation
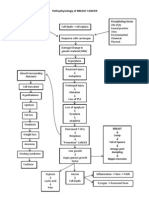
- Pathophysiology of BREAST CANCERDocument1 pagePathophysiology of BREAST CANCERAlinor Abubacar100% (6)
- Fermentation Process KineticsDocument7 pagesFermentation Process KineticsDillip_subuPas encore d'évaluation
- Root Resorption: Challenge To The Endodontist: Acta Scientific Dental SciencesDocument11 pagesRoot Resorption: Challenge To The Endodontist: Acta Scientific Dental SciencesMuhammadDeniRahmanPas encore d'évaluation
- Seminar Hydrocephalus Surgery Yr 4 Rotation 3Document81 pagesSeminar Hydrocephalus Surgery Yr 4 Rotation 3BorhanPas encore d'évaluation
- Practical 14 PDFDocument3 pagesPractical 14 PDFSiddharth KumraPas encore d'évaluation
- Hematopoietic Agents and Hematinics: PharmacologyDocument9 pagesHematopoietic Agents and Hematinics: PharmacologyAbi SulitPas encore d'évaluation
- 4 - Subphylum UrochordataDocument12 pages4 - Subphylum UrochordataStudent 365Pas encore d'évaluation
- DK Guide To The Human Body PDFDocument67 pagesDK Guide To The Human Body PDFTina Fishie Volf100% (5)
- Pathologyofdiseasesof Geriatricexoticmammals: Drury R. Reavill,, Denise M. ImaiDocument34 pagesPathologyofdiseasesof Geriatricexoticmammals: Drury R. Reavill,, Denise M. ImaiRaquel MotaPas encore d'évaluation
- 1.13.2 Clinical Localization and History in NeurologyDocument42 pages1.13.2 Clinical Localization and History in Neurologyfikrah sharifPas encore d'évaluation
- Causes and prevention of cancerDocument4 pagesCauses and prevention of cancerosamaPas encore d'évaluation
- FTND - Harmful Effects of Pornography - s-10915 PDFDocument100 pagesFTND - Harmful Effects of Pornography - s-10915 PDFponderatulPas encore d'évaluation
- BugSpeaks Sample ReportDocument27 pagesBugSpeaks Sample ReportAnonymous 76wOMTpPas encore d'évaluation
- Congenital Heart Disease SatpathyDocument383 pagesCongenital Heart Disease SatpathyTanvir AhmedPas encore d'évaluation