Vous aimerez peut-être aussi
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Presentation5 EV ArchitectureDocument26 pagesPresentation5 EV ArchitectureJAYKUMAR MUKESHBHAI THAKORPas encore d'évaluation
- Commissioning PFDC DriveDocument45 pagesCommissioning PFDC DriveFonoaudióloga Gisele RezendePas encore d'évaluation
- Bent's RuleDocument3 pagesBent's RuleEdwinPas encore d'évaluation
- IncarnationDocument3 pagesIncarnationViviana PueblaPas encore d'évaluation
- Data Download CMM366A-4G V1.0 enDocument16 pagesData Download CMM366A-4G V1.0 enSuramanPas encore d'évaluation
- Module 1 Engineering ScienceDocument38 pagesModule 1 Engineering ScienceLogan JessePas encore d'évaluation
- en Product OverviewDocument81 pagesen Product OverviewShakeel AhmedPas encore d'évaluation
- Baulkham Hills 2012 2U Prelim Yearly & SolutionsDocument11 pagesBaulkham Hills 2012 2U Prelim Yearly & SolutionsYe ZhangPas encore d'évaluation
- Python Unit 1Document18 pagesPython Unit 1Rtr. Venkata chetan Joint secretaryPas encore d'évaluation
- Differential Pr. Gauges Bellow Type 1Document2 pagesDifferential Pr. Gauges Bellow Type 1Vara PrasadPas encore d'évaluation
- Form 1 ExercisesDocument160 pagesForm 1 Exerciseskays MPas encore d'évaluation
- Computational Neuroscience CW1 (Bristol)Document3 pagesComputational Neuroscience CW1 (Bristol)libannfPas encore d'évaluation
- V1 N2 1980 RabenhorstDocument6 pagesV1 N2 1980 Rabenhorstraa2010Pas encore d'évaluation
- Physics Sample Question PaperDocument9 pagesPhysics Sample Question PaperVarsha SharmaPas encore d'évaluation
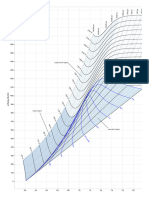
- Mollier Enthalpy Entropy Chart For Steam - US UnitsDocument1 pageMollier Enthalpy Entropy Chart For Steam - US Unitslin tongPas encore d'évaluation
- Ee-502 Unit - IDocument2 pagesEe-502 Unit - IVARAPRASADPas encore d'évaluation
- 2010 Jan-01Document32 pages2010 Jan-01Shine PrabhakaranPas encore d'évaluation
- List of Books s8 ElectiveDocument2 pagesList of Books s8 ElectivemaniPas encore d'évaluation
- International Marketing-Assignment No. 01Document36 pagesInternational Marketing-Assignment No. 01Faisal Shahzad60% (5)
- PLX7100A Digital Mobile C-Arm X-Ray Machine: 1. Technical SpecificationDocument3 pagesPLX7100A Digital Mobile C-Arm X-Ray Machine: 1. Technical SpecificationAbdalhakeem AlturkyPas encore d'évaluation
- Spesiikasi PerallatanDocument10 pagesSpesiikasi PerallatanRafi RaziqPas encore d'évaluation
- 6100 SQ Lcms Data SheetDocument4 pages6100 SQ Lcms Data Sheet王皓Pas encore d'évaluation
- Newvhdl Syllabus (It&Cse)Document2 pagesNewvhdl Syllabus (It&Cse)Mude Kishore NaikPas encore d'évaluation
- Mathematical Analysisand Optimizationfor EconomistsDocument4 pagesMathematical Analysisand Optimizationfor EconomistsGuillermo GómezPas encore d'évaluation
- Magnetic Field of A SolenoidDocument5 pagesMagnetic Field of A SolenoidKang Yuan ShingPas encore d'évaluation
- Prosper & Sucker RodDocument20 pagesProsper & Sucker RodOmar AbdoPas encore d'évaluation
- Switching Power Supply DesignDocument21 pagesSwitching Power Supply DesignSamuel mutindaPas encore d'évaluation
- C.KESAVAN - Diploma EEE: Phone No Mail IdDocument3 pagesC.KESAVAN - Diploma EEE: Phone No Mail IdKesavan ChinaswmiPas encore d'évaluation
- 16 - Bit RISC Processor Design For Convolution Application Using Verilog HDLDocument64 pages16 - Bit RISC Processor Design For Convolution Application Using Verilog HDLchandra sekhar100% (1)
- A B C D: Choose Only One Answer For Each QuestionDocument10 pagesA B C D: Choose Only One Answer For Each QuestionAchitt AchitPas encore d'évaluation