Vous aimerez peut-être aussi
- Best Practices of Apheresis in Hematopoietic Cell TransplantationD'EverandBest Practices of Apheresis in Hematopoietic Cell TransplantationSyed A. AbutalibPas encore d'évaluation
- Diagnosis and Management of Paroxysmal Nocturnal HemoglobinuriaDocument11 pagesDiagnosis and Management of Paroxysmal Nocturnal Hemoglobinuriaida ayu agung WijayantiPas encore d'évaluation
- Diagnosis of Blood and Bone Marrow DisordersD'EverandDiagnosis of Blood and Bone Marrow DisordersSa A. WangPas encore d'évaluation
- Diagnosis and Management of PNHDocument12 pagesDiagnosis and Management of PNHapi-26302710Pas encore d'évaluation
- Diagnostic Pathology of Hematopoietic Disorders of Spleen and LiverD'EverandDiagnostic Pathology of Hematopoietic Disorders of Spleen and LiverPas encore d'évaluation
- NIH Public Access: Author ManuscriptDocument17 pagesNIH Public Access: Author ManuscriptyostiPas encore d'évaluation
- Clinical Cases in Heart FailureD'EverandClinical Cases in Heart FailureRavi V. ShahPas encore d'évaluation
- 291.full Anomalia Pelger Huet PseudoDocument13 pages291.full Anomalia Pelger Huet PseudoEvitaPumitaPas encore d'évaluation
- How I Treat Acquired Aplastic AnemiaDocument13 pagesHow I Treat Acquired Aplastic AnemiaJohn chuksPas encore d'évaluation
- Treatment For Aplastic AnemiaDocument13 pagesTreatment For Aplastic Anemiagustianto hutama pPas encore d'évaluation
- Cytometry Part B Clinical - 2017 - Dezern - ICCS ESCCA Consensus Guidelines To Detect GPI Deficient Cells in ParoxysmalDocument8 pagesCytometry Part B Clinical - 2017 - Dezern - ICCS ESCCA Consensus Guidelines To Detect GPI Deficient Cells in ParoxysmalzafeersmaPas encore d'évaluation
- Aplastic Anemia - An Overview: DR Aniruddh Shrivastava Guided By: DR S.H. Talib SIRDocument42 pagesAplastic Anemia - An Overview: DR Aniruddh Shrivastava Guided By: DR S.H. Talib SIRdoctoranswerit_84161Pas encore d'évaluation
- Prevalence of Anemia in Patients With Heart Failure: Edition: 29.1 - 12 Article(s)Document13 pagesPrevalence of Anemia in Patients With Heart Failure: Edition: 29.1 - 12 Article(s)Novia KurniantiPas encore d'évaluation
- Low Blood Counts: Immune Mediated, Idiopathic, or MyelodysplasiaDocument7 pagesLow Blood Counts: Immune Mediated, Idiopathic, or MyelodysplasiaaaaaPas encore d'évaluation
- AAFP Marzo 2016 Sindrome NefroticoDocument7 pagesAAFP Marzo 2016 Sindrome NefroticoAntonio MoncadaPas encore d'évaluation
- Doruk Erkan Expert Perspective Management ofDocument26 pagesDoruk Erkan Expert Perspective Management ofSrinivas PingaliPas encore d'évaluation
- Anemia ThesisDocument4 pagesAnemia ThesisFinni Rice100% (2)
- Assignment 1 Pathology Aplastic Anemia: Supervisor: DR - Ramez Al-KeelaniDocument6 pagesAssignment 1 Pathology Aplastic Anemia: Supervisor: DR - Ramez Al-Keelaniameer mousaPas encore d'évaluation
- Pseudo Pelger Huet Anomaly Induced by MedicationsDocument13 pagesPseudo Pelger Huet Anomaly Induced by MedicationsHam BonePas encore d'évaluation
- Paroxysmal Nocturnal Hemoglobinuria Testing in Patients With Myelodysplastic Syndrome in Clinical PracticeDocument7 pagesParoxysmal Nocturnal Hemoglobinuria Testing in Patients With Myelodysplastic Syndrome in Clinical PracticeNancy Fanny Vega ZuñigaPas encore d'évaluation
- Evaluationandmanagement Ofprimary Hyperaldosteronism: Frances T. Lee,, Dina ElarajDocument15 pagesEvaluationandmanagement Ofprimary Hyperaldosteronism: Frances T. Lee,, Dina ElarajDiego BallesterosPas encore d'évaluation
- Advancesadv 2021004345Document12 pagesAdvancesadv 2021004345Ari Dwi PrasetyoPas encore d'évaluation
- The Recommendations of A Consensus Panel For The Screening, Diagnosis, and Treatment of Neurogenic Orthostatic Hypotension and Associated Supine Hypertension (Journal of Neurology, 2017)Document16 pagesThe Recommendations of A Consensus Panel For The Screening, Diagnosis, and Treatment of Neurogenic Orthostatic Hypotension and Associated Supine Hypertension (Journal of Neurology, 2017)mysticmdPas encore d'évaluation
- Aplastic AnemiaDocument29 pagesAplastic AnemiaAshish SoniPas encore d'évaluation
- 10.1515 - Labmed 2016 0017Document16 pages10.1515 - Labmed 2016 0017baniamerabdullah88Pas encore d'évaluation
- Go 2006Document12 pagesGo 2006my accountPas encore d'évaluation
- Approach Considerations: Magnetic Resonance Imaging in Research SettingsDocument5 pagesApproach Considerations: Magnetic Resonance Imaging in Research SettingsLamyaa Ali HasanPas encore d'évaluation
- Aplastic Anemia - Pathogenesis, Clinical Manifestations, and Diagnosis - UpToDate PDFDocument21 pagesAplastic Anemia - Pathogenesis, Clinical Manifestations, and Diagnosis - UpToDate PDFWahyu Dwi NugrohoPas encore d'évaluation
- ANEMIA P ('t':3) Var B Location Settimeout (Function (If (Typeof Window - Iframe 'Undefined') (B.href B.href ) ), 15000)Document7 pagesANEMIA P ('t':3) Var B Location Settimeout (Function (If (Typeof Window - Iframe 'Undefined') (B.href B.href ) ), 15000)Arv IraPas encore d'évaluation
- Advancesadv 2021004345Document12 pagesAdvancesadv 2021004345No NamePas encore d'évaluation
- Impairment of Bone Health in Pediatric Patients With Hemolytic AnemiaDocument7 pagesImpairment of Bone Health in Pediatric Patients With Hemolytic AnemiaMarifer EstradaPas encore d'évaluation
- Jurnal Anemia Aplastik PDFDocument28 pagesJurnal Anemia Aplastik PDFniaasetaPas encore d'évaluation
- Anemia in Heart Failure: Pathophysiology, Pathogenesis, Treatment, and IncognitaeDocument13 pagesAnemia in Heart Failure: Pathophysiology, Pathogenesis, Treatment, and IncognitaeHedya Nadhrati SururaPas encore d'évaluation
- EPO in Anemia CHFDocument10 pagesEPO in Anemia CHFAlizaPinkyPas encore d'évaluation
- Thrombotic Thrombocytopenic Purpura and Hemolytic Uremic SyndromeDocument9 pagesThrombotic Thrombocytopenic Purpura and Hemolytic Uremic SyndromeRex RuthorPas encore d'évaluation
- Clinico - Hematological Analysis of Pancytopenia: A Bone Marrow StudyDocument6 pagesClinico - Hematological Analysis of Pancytopenia: A Bone Marrow StudySrinath M VPas encore d'évaluation
- 55421-Gagal JantungDocument21 pages55421-Gagal JantungAngie CouthalooPas encore d'évaluation
- Hemangioma InmunohistoquimicaDocument20 pagesHemangioma InmunohistoquimicaSalome ChaconPas encore d'évaluation
- An Etiological Reappraisal of Pancytopenia - LargestDocument9 pagesAn Etiological Reappraisal of Pancytopenia - LargestKaye Antonette AntioquiaPas encore d'évaluation
- Nihms 1813067Document34 pagesNihms 1813067Marlyn SuciningtiasPas encore d'évaluation
- Cakmakli 2018Document8 pagesCakmakli 2018dora guzmanPas encore d'évaluation
- Anemias Aplastic Anemia Is A Condition Where Bone Marrow Does Not Produce Sufficient NewDocument15 pagesAnemias Aplastic Anemia Is A Condition Where Bone Marrow Does Not Produce Sufficient NewZoreyca RiveraPas encore d'évaluation
- Summary of Summary 5Document36 pagesSummary of Summary 5anon-265120100% (2)
- Prevalence of Ventricular Arrhythmia and Its Associated Factors in Nondialyzed Chronic Kidney Disease PatientsDocument8 pagesPrevalence of Ventricular Arrhythmia and Its Associated Factors in Nondialyzed Chronic Kidney Disease Patientsjustin_sanePas encore d'évaluation
- Jurnal 6Document21 pagesJurnal 6onisPas encore d'évaluation
- Autosomal Recessive Hypercholesterolemia: Case ReportDocument7 pagesAutosomal Recessive Hypercholesterolemia: Case ReportLADY DIANEE SOTOMAYOR GUTIERREZPas encore d'évaluation
- American J Hematol - 2023 - PanseDocument14 pagesAmerican J Hematol - 2023 - PanseMaria GarciaPas encore d'évaluation
- Hepatic Encephalopathy Novel Insights Into Classification, Pathophysiology and TherapyDocument22 pagesHepatic Encephalopathy Novel Insights Into Classification, Pathophysiology and TherapyMichelle RdgzPas encore d'évaluation
- In Practice Blood Transfusion in Dogs and Cats1Document7 pagesIn Practice Blood Transfusion in Dogs and Cats1何元Pas encore d'évaluation
- Ehf2 8 625Document9 pagesEhf2 8 625elza pratiwiPas encore d'évaluation
- Biruh T Workeneh Hyponatremia Demystified IntegratingDocument17 pagesBiruh T Workeneh Hyponatremia Demystified IntegratingsaqqarazoserPas encore d'évaluation
- Penugasan - Diagnosis and Treatment of Aplastic AnemiaDocument22 pagesPenugasan - Diagnosis and Treatment of Aplastic AnemiaokerPas encore d'évaluation
- Postdialysis Hypertension: Associated Factors, Patient Profiles, and Cardiovascular MortalityDocument6 pagesPostdialysis Hypertension: Associated Factors, Patient Profiles, and Cardiovascular MortalityLaura PutriPas encore d'évaluation
- Mini-Review Heavy Menstrual Bleeding As A Common Presenting Symptom of Rare Platelet Disorders: Illustrative Case ExamplesDocument5 pagesMini-Review Heavy Menstrual Bleeding As A Common Presenting Symptom of Rare Platelet Disorders: Illustrative Case Examplesimmanuel dioPas encore d'évaluation
- Aplastic Anemia: Review of Etiology and TreatmentDocument7 pagesAplastic Anemia: Review of Etiology and TreatmentCleber MaiaPas encore d'évaluation
- Noncirrhotic Portal Hypertension: ReviewDocument6 pagesNoncirrhotic Portal Hypertension: ReviewIsrael BlancoPas encore d'évaluation
- Thrombotic Thrombocytopenic PurpuraDocument5 pagesThrombotic Thrombocytopenic PurpuraAhsan Tanio DaulayPas encore d'évaluation
- Update On (Approach To) Anemia1 (Changes)Document39 pagesUpdate On (Approach To) Anemia1 (Changes)Balchand KukrejaPas encore d'évaluation
- Paroxysmal Nocturnal Hemoglobinuria: March 8, 2005Document46 pagesParoxysmal Nocturnal Hemoglobinuria: March 8, 2005Farina ReenaPas encore d'évaluation
- Antiphospholipid Thrombosis Syndrome HematoFeb2008, Vol. 22Document173 pagesAntiphospholipid Thrombosis Syndrome HematoFeb2008, Vol. 22aazizkhanPas encore d'évaluation
- Lung Carcinoma: Radiotherapy PlanningDocument9 pagesLung Carcinoma: Radiotherapy PlanningTowhidulIslamPas encore d'évaluation
- Carcinoma Rectum - Janak - NEWDocument74 pagesCarcinoma Rectum - Janak - NEWTowhidulIslamPas encore d'évaluation
- Gastric Carcinoma: Radiation Planning: Ebrt Assessment of Disease 1. Examinations: 2. Investigations: A. Non-InvasiveDocument33 pagesGastric Carcinoma: Radiation Planning: Ebrt Assessment of Disease 1. Examinations: 2. Investigations: A. Non-InvasiveTowhidulIslamPas encore d'évaluation
- 0529 Protocol Update 6.2.09 PDFDocument57 pages0529 Protocol Update 6.2.09 PDFTowhidulIslamPas encore d'évaluation
- Adult Body Mass Index (BMI) Chart: WeightDocument2 pagesAdult Body Mass Index (BMI) Chart: WeightTowhidulIslamPas encore d'évaluation
- Dr. Towhidul Islam: MBBS, BCS (Health) CCD Bangabandhu Sheikh Mujib Medical University Cell Phone: 01725271380Document1 pageDr. Towhidul Islam: MBBS, BCS (Health) CCD Bangabandhu Sheikh Mujib Medical University Cell Phone: 01725271380TowhidulIslamPas encore d'évaluation
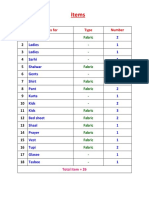
- Items: No Items For Type NumberDocument1 pageItems: No Items For Type NumberTowhidulIslamPas encore d'évaluation
- Carcinoma Rectum - Janak - NEWDocument74 pagesCarcinoma Rectum - Janak - NEWTowhidulIslamPas encore d'évaluation
- Horton 2018Document5 pagesHorton 2018TowhidulIslamPas encore d'évaluation
- Pregnancy Fact SheetDocument3 pagesPregnancy Fact SheetTowhidulIslamPas encore d'évaluation
- Fade To Black: Ride The LightningDocument7 pagesFade To Black: Ride The LightningTowhidulIslamPas encore d'évaluation
- Abvd Hem HLDocument6 pagesAbvd Hem HLTowhidulIslamPas encore d'évaluation
- Ema-Co Gy GTDDocument4 pagesEma-Co Gy GTDTowhidulIslamPas encore d'évaluation
- Floppy BabyDocument13 pagesFloppy BabyJorge JhgPas encore d'évaluation
- Manajemen Nyeri Dengan DexketoprofenDocument27 pagesManajemen Nyeri Dengan Dexketoprofenmaya santiPas encore d'évaluation
- A Client With A Brain Tumor: Nursing Care PlanDocument1 pageA Client With A Brain Tumor: Nursing Care Planshabatat2002Pas encore d'évaluation
- Copia Di Drugs That Cause or Prolong Delirium - UpToDateDocument2 pagesCopia Di Drugs That Cause or Prolong Delirium - UpToDateAFA.BLSPas encore d'évaluation
- NehaDocument20 pagesNehaRecorders RndPas encore d'évaluation
- Drug StudyDocument21 pagesDrug StudyShyla Garnace JavillonarPas encore d'évaluation
- Childrens Global Assessment of Functioning ScaleDocument1 pageChildrens Global Assessment of Functioning Scalezb789100% (1)
- Columbine Report Pgs 9201-9300Document100 pagesColumbine Report Pgs 9201-9300columbinefamilyrequestPas encore d'évaluation
- Unit 5-Heart QuestionsDocument37 pagesUnit 5-Heart Questionsareyouthere92100% (1)
- Speaking Short QuestionDocument8 pagesSpeaking Short QuestionpraveshkaflePas encore d'évaluation
- SdasdasdasdasdDocument4 pagesSdasdasdasdasdRadu MelnicPas encore d'évaluation
- WarmTouch User Manual Wt5200 UsDocument34 pagesWarmTouch User Manual Wt5200 Usnobel0001Pas encore d'évaluation
- Cardiology PDFDocument8 pagesCardiology PDFTran Chi TienPas encore d'évaluation
- Health Assessment in NursingDocument48 pagesHealth Assessment in NursingCj MayoyoPas encore d'évaluation
- Bmi Data May Joie J. AlcarazDocument13 pagesBmi Data May Joie J. AlcarazMay Joie JaymePas encore d'évaluation
- Aversion TherapyDocument11 pagesAversion Therapyayyappan.ashok6713100% (2)
- Housekeeping Check Lists-UpdatedDocument26 pagesHousekeeping Check Lists-UpdatedtanishaPas encore d'évaluation
- Waukboard Sellsheet v13Document1 pageWaukboard Sellsheet v13Forum PompieriiPas encore d'évaluation
- Final FinalDocument10 pagesFinal FinalSimeonDelaRosaPas encore d'évaluation
- HANDOUTDocument2 pagesHANDOUT27 Nguyễn Thị Huyền TrangPas encore d'évaluation
- Suppositories CalculationsDocument9 pagesSuppositories CalculationsYuppie Raj67% (3)
- Admission NUHDocument2 pagesAdmission NUHDanny WidjajaPas encore d'évaluation
- Anticoagulation in EcmoDocument13 pagesAnticoagulation in Ecmoapi-534214500Pas encore d'évaluation
- Consumos Medicamentos 2020Document568 pagesConsumos Medicamentos 2020LUCERO LOPEZPas encore d'évaluation
- Dka CalculatorDocument1 pageDka CalculatordelfiaPas encore d'évaluation
- Blood Composition For REPRODUCEDocument2 pagesBlood Composition For REPRODUCEJasminePas encore d'évaluation
- Patanjali Case StudyDocument7 pagesPatanjali Case Studysurz_emperorPas encore d'évaluation
- Case Presentation On LrtiDocument17 pagesCase Presentation On LrtiNewtan DebPas encore d'évaluation
- AsafoetidaDocument2 pagesAsafoetidameet143bmPas encore d'évaluation
- Arnica The Miracle Remedy - Case RecordsDocument4 pagesArnica The Miracle Remedy - Case Recordskaravi schiniasPas encore d'évaluation
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)D'EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Évaluation : 3 sur 5 étoiles3/5 (1)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDD'EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDÉvaluation : 5 sur 5 étoiles5/5 (4)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionD'EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionÉvaluation : 4 sur 5 étoiles4/5 (404)
- The Comfort of Crows: A Backyard YearD'EverandThe Comfort of Crows: A Backyard YearÉvaluation : 4.5 sur 5 étoiles4.5/5 (23)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsD'EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsÉvaluation : 4 sur 5 étoiles4/5 (4)
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedD'EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedÉvaluation : 4.5 sur 5 étoiles4.5/5 (82)
- The Age of Magical Overthinking: Notes on Modern IrrationalityD'EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityÉvaluation : 4 sur 5 étoiles4/5 (34)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryD'EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryÉvaluation : 4 sur 5 étoiles4/5 (46)
- The Obesity Code: Unlocking the Secrets of Weight LossD'EverandThe Obesity Code: Unlocking the Secrets of Weight LossÉvaluation : 4 sur 5 étoiles4/5 (6)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsD'EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsPas encore d'évaluation
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeD'EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeÉvaluation : 2 sur 5 étoiles2/5 (1)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsD'EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsÉvaluation : 4.5 sur 5 étoiles4.5/5 (170)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaD'EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsD'EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsÉvaluation : 5 sur 5 étoiles5/5 (1)
- The Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeD'EverandThe Twentysomething Treatment: A Revolutionary Remedy for an Uncertain AgeÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)
- Manipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesD'EverandManipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesÉvaluation : 4.5 sur 5 étoiles4.5/5 (1412)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisD'EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisÉvaluation : 4.5 sur 5 étoiles4.5/5 (44)
- Summary: Limitless: Upgrade Your Brain, Learn Anything Faster, and Unlock Your Exceptional Life By Jim Kwik: Key Takeaways, Summary and AnalysisD'EverandSummary: Limitless: Upgrade Your Brain, Learn Anything Faster, and Unlock Your Exceptional Life By Jim Kwik: Key Takeaways, Summary and AnalysisÉvaluation : 5 sur 5 étoiles5/5 (8)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.D'EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Évaluation : 4.5 sur 5 étoiles4.5/5 (110)
- How to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingD'EverandHow to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingÉvaluation : 1 sur 5 étoiles1/5 (2)
- To Explain the World: The Discovery of Modern ScienceD'EverandTo Explain the World: The Discovery of Modern ScienceÉvaluation : 3.5 sur 5 étoiles3.5/5 (51)
- Self-Care for Autistic People: 100+ Ways to Recharge, De-Stress, and Unmask!D'EverandSelf-Care for Autistic People: 100+ Ways to Recharge, De-Stress, and Unmask!Évaluation : 5 sur 5 étoiles5/5 (1)
- The Garden Within: Where the War with Your Emotions Ends and Your Most Powerful Life BeginsD'EverandThe Garden Within: Where the War with Your Emotions Ends and Your Most Powerful Life BeginsPas encore d'évaluation
- Critical Thinking: How to Effectively Reason, Understand Irrationality, and Make Better DecisionsD'EverandCritical Thinking: How to Effectively Reason, Understand Irrationality, and Make Better DecisionsÉvaluation : 4.5 sur 5 étoiles4.5/5 (39)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeD'EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeÉvaluation : 4.5 sur 5 étoiles4.5/5 (254)