Vous aimerez peut-être aussi
- Faculty of Business - Report WritingDocument16 pagesFaculty of Business - Report WritingcuambyahooPas encore d'évaluation
- Advt - No - 1-2018Document5 pagesAdvt - No - 1-2018aminaPas encore d'évaluation
- Faculty of Business - Report WritingDocument16 pagesFaculty of Business - Report WritingcuambyahooPas encore d'évaluation
- Peterdruckers Whatmakesaneffectiveleaderpps 150823134802 Lva1 App6891Document20 pagesPeterdruckers Whatmakesaneffectiveleaderpps 150823134802 Lva1 App6891cuambyahooPas encore d'évaluation
- PPSC Advt 60-2017 - 48cmx8colDocument1 pagePPSC Advt 60-2017 - 48cmx8colcuambyahooPas encore d'évaluation
- Pharmacy Act 67Document11 pagesPharmacy Act 67Aqeel AhmedPas encore d'évaluation
- Procurement Cycle PDFDocument1 pageProcurement Cycle PDFcuambyahoo100% (1)
- Medialeer Engels HsDocument1 pageMedialeer Engels HscuambyahooPas encore d'évaluation
- Advt - No - 1-2018Document5 pagesAdvt - No - 1-2018aminaPas encore d'évaluation
- Quetta Attack PDFDocument1 pageQuetta Attack PDFcuambyahooPas encore d'évaluation
- Balance Sheet Format PDFDocument1 pageBalance Sheet Format PDFpraveenyarandolePas encore d'évaluation
- Pharmacy Act 67Document11 pagesPharmacy Act 67Aqeel AhmedPas encore d'évaluation
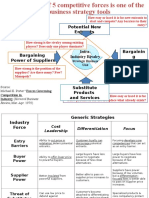
- Potential New Entrants: Strategic Business UnitDocument6 pagesPotential New Entrants: Strategic Business UnitcuambyahooPas encore d'évaluation
- HomeostasisDocument36 pagesHomeostasisUwais AhmedPas encore d'évaluation
- Immigration To Australia-StepbyStep Guide (Subclass 189&190) V3.0Document5 pagesImmigration To Australia-StepbyStep Guide (Subclass 189&190) V3.0cuambyahooPas encore d'évaluation
- Schmal Cdncrimtdy 2e Ch11 (Compatibility Mode)Document2 pagesSchmal Cdncrimtdy 2e Ch11 (Compatibility Mode)cuambyahooPas encore d'évaluation
- Schmal - Cdncrimtdy - 2e - ch03 (Compatibility Mode) PDFDocument2 pagesSchmal - Cdncrimtdy - 2e - ch03 (Compatibility Mode) PDFcuambyahooPas encore d'évaluation
- Quetta Attack PDFDocument1 pageQuetta Attack PDFcuambyahooPas encore d'évaluation
- Democracy Accountability and RepresentationDocument25 pagesDemocracy Accountability and RepresentationcuambyahooPas encore d'évaluation
- Aleppo massacres and need for justiceDocument28 pagesAleppo massacres and need for justicecuambyahooPas encore d'évaluation
- Workshop IntroDocument41 pagesWorkshop Introjdpatel28Pas encore d'évaluation
- Richard Nixon - Resignation Address PDFDocument4 pagesRichard Nixon - Resignation Address PDFcuambyahooPas encore d'évaluation
- Enthalpy S&G 06Document13 pagesEnthalpy S&G 06OnSolomonPas encore d'évaluation
- Impact of Demographic Changes On Inflation in Pakistan: A A J, F F F MDocument1 pageImpact of Demographic Changes On Inflation in Pakistan: A A J, F F F McuambyahooPas encore d'évaluation
- Criminology Reviewer For Licensure Exam And Board ExaminationDocument48 pagesCriminology Reviewer For Licensure Exam And Board ExaminationJona Addatu96% (24)
- A Kashmir Statement by Ashraf JhangirDocument5 pagesA Kashmir Statement by Ashraf JhangircuambyahooPas encore d'évaluation
- Minority Report 2016Document70 pagesMinority Report 2016cuambyahooPas encore d'évaluation
- 17 LectureDocument61 pages17 LecturecuambyahooPas encore d'évaluation
- Area Population Density and Urban Rural Proportion, PakistanDocument1 pageArea Population Density and Urban Rural Proportion, PakistanZeibJahangirPas encore d'évaluation
- Pakistan 1998 Census Unemployment Rates by Gender and LocationDocument1 pagePakistan 1998 Census Unemployment Rates by Gender and LocationcuambyahooPas encore d'évaluation
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5782)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (890)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (72)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- AY5110 Auditing & Assurance I & AY516 - Sample Paper 2017 - 2018Document7 pagesAY5110 Auditing & Assurance I & AY516 - Sample Paper 2017 - 2018Priya TomarPas encore d'évaluation
- Step 3: Architect Your Success: Define Your Process FlowDocument7 pagesStep 3: Architect Your Success: Define Your Process Flowhervin mamaniPas encore d'évaluation
- Case Analysis - Charlotte Beers at Ogilvy and Mather WorldwideDocument6 pagesCase Analysis - Charlotte Beers at Ogilvy and Mather WorldwideNaheed Khan100% (2)
- BÁO CÁO THỰC TẬP - TRẦN THỊ THÙY DƯƠNG - 7567Document22 pagesBÁO CÁO THỰC TẬP - TRẦN THỊ THÙY DƯƠNG - 7567Nicolette TuanPas encore d'évaluation
- Pledge: Coverage of Discussion: Kinds of Pledge Conventional Pledge Legal PledgeDocument26 pagesPledge: Coverage of Discussion: Kinds of Pledge Conventional Pledge Legal PledgeAmie Jane MirandaPas encore d'évaluation
- Oromiya Cooperative BankDocument18 pagesOromiya Cooperative BankMIKIYASPas encore d'évaluation
- Full Package - New Driver Aplication PDFDocument11 pagesFull Package - New Driver Aplication PDFdina junu100% (1)
- Study On: A Project Report OnDocument20 pagesStudy On: A Project Report OnVIVEK MEHTAPas encore d'évaluation
- Accrual Basis Accounting IllustratedDocument2 pagesAccrual Basis Accounting IllustratedAye TubePas encore d'évaluation
- ACT 311 - A - 12018001086 - Jason SaputeraDocument6 pagesACT 311 - A - 12018001086 - Jason Saputerajuljul julioPas encore d'évaluation
- Effective Resume: The First Step Toward Landing Your JobDocument23 pagesEffective Resume: The First Step Toward Landing Your JobDhaval VikamseyPas encore d'évaluation
- Essai - Officialstatistic - 19611155 - M.paris Ramdoni Rasantaka - DDocument6 pagesEssai - Officialstatistic - 19611155 - M.paris Ramdoni Rasantaka - DParis RamdaniPas encore d'évaluation
- Python Binance Readthedocs Io en StableDocument226 pagesPython Binance Readthedocs Io en StableAndres1969Pas encore d'évaluation
- Abhishek Verma - MBA FinanceDocument17 pagesAbhishek Verma - MBA FinancePragati ChaudharyPas encore d'évaluation
- MHTP 2Document2 pagesMHTP 2Nabeel MohammadPas encore d'évaluation
- FRA AssignmentDocument12 pagesFRA AssignmentsomechnitjPas encore d'évaluation
- Tax 2 - DST, ExciseDocument11 pagesTax 2 - DST, ExciseDINARDO SANTOSPas encore d'évaluation
- Sample Kaizen Paper 1 - FIVEGENDocument8 pagesSample Kaizen Paper 1 - FIVEGENerilPas encore d'évaluation
- Mattly v. Brown/focus, Ariz. Ct. App. (2016)Document8 pagesMattly v. Brown/focus, Ariz. Ct. App. (2016)Scribd Government DocsPas encore d'évaluation
- MCD FranchiseDocument10 pagesMCD FranchiseLastri Anggi FaniPas encore d'évaluation
- Salary Sheet Format (Latest)Document5 pagesSalary Sheet Format (Latest)AMIT SHARMAPas encore d'évaluation
- SGTDocument10 pagesSGTFlowSikk -Pas encore d'évaluation
- Strategic Entrepreneurship: Fourth Edition © 2007 Philip A WickhamDocument14 pagesStrategic Entrepreneurship: Fourth Edition © 2007 Philip A WickhamerikloevgrenPas encore d'évaluation
- The Story Triangle PDFDocument20 pagesThe Story Triangle PDFabcgoa50% (4)
- Viñalon, Angelica V. (Entrepreneurial Mind, Module 1)Document5 pagesViñalon, Angelica V. (Entrepreneurial Mind, Module 1)Angelica Villaflor ViñalonPas encore d'évaluation
- Muhammad Tughlaq's Token CurrencyDocument3 pagesMuhammad Tughlaq's Token CurrencyAnjanaPas encore d'évaluation
- Charms and ChallengesDocument11 pagesCharms and ChallengesOm PrasadPas encore d'évaluation
- GIST Innovation HubDocument24 pagesGIST Innovation Hubtony_553091275Pas encore d'évaluation
- Electricity Bill ExplainedDocument1 pageElectricity Bill ExplainedCliff Mokua100% (3)
- 1 - SAP Extended Warehouse ManagementDocument45 pages1 - SAP Extended Warehouse ManagementMahesh Mokashi100% (2)