Vous aimerez peut-être aussi
- Articulo LisisDocument8 pagesArticulo LisisAle DomínguezPas encore d'évaluation
- Epidemiological Study of Clinical and Laboratory Profiles of Patients Woth ALL at DR SoetomoDocument5 pagesEpidemiological Study of Clinical and Laboratory Profiles of Patients Woth ALL at DR SoetomoJulia Intan Permata SariPas encore d'évaluation
- Acute Fatty Liver of Pregnancy: Clinical Features and Treatment Strategies in A Single-Center StudyDocument3 pagesAcute Fatty Liver of Pregnancy: Clinical Features and Treatment Strategies in A Single-Center StudyIJAR JOURNALPas encore d'évaluation
- Central Nervous System Tumours of ChildhoodD'EverandCentral Nervous System Tumours of ChildhoodEdward EstlinPas encore d'évaluation
- A Double-Blind, Placebo-Controlled Trial of Ruxolitinib For MyelofibrosisDocument9 pagesA Double-Blind, Placebo-Controlled Trial of Ruxolitinib For MyelofibrosisharlessitompulPas encore d'évaluation
- 1 s2.0 S1198743X14610478 MainDocument17 pages1 s2.0 S1198743X14610478 Mainazkarazkar2001Pas encore d'évaluation
- Literature Review of Chronic Liver DiseaseDocument6 pagesLiterature Review of Chronic Liver Diseaseafmzodjhpxembt100% (1)
- Tatalaksana Abses HeparDocument6 pagesTatalaksana Abses HeparAdhi Rizky PutraPas encore d'évaluation
- Management of Occult GI Bleeding: A Clinical GuideD'EverandManagement of Occult GI Bleeding: A Clinical GuideMicheal TadrosPas encore d'évaluation
- Art GEBADocument8 pagesArt GEBAJhonathan Andres Garcia FiallosPas encore d'évaluation
- Evaluation and Management of Dysphagia: An Evidence-Based ApproachD'EverandEvaluation and Management of Dysphagia: An Evidence-Based ApproachDhyanesh A. PatelPas encore d'évaluation
- Liver Transplantation: Challenging Controversies and TopicsD'EverandLiver Transplantation: Challenging Controversies and TopicsPas encore d'évaluation
- Absceso Hepatico PiogenoDocument6 pagesAbsceso Hepatico PiogenoPablo Eduardo AmadorPas encore d'évaluation
- Essential Medical Disorders of the Stomach and Small Intestine: A Clinical CasebookD'EverandEssential Medical Disorders of the Stomach and Small Intestine: A Clinical CasebookPas encore d'évaluation
- Rare Diseases and Orphan Drugs: Keys to Understanding and Treating the Common DiseasesD'EverandRare Diseases and Orphan Drugs: Keys to Understanding and Treating the Common DiseasesPas encore d'évaluation
- Development of Unfavorable Hepatoblastoma in Children of Very Low Birth WeightDocument8 pagesDevelopment of Unfavorable Hepatoblastoma in Children of Very Low Birth WeightKevinDilianSugandaPas encore d'évaluation
- Awareness of Patients With Symptomatic Gallstones Regarding Their Own DiseaseDocument3 pagesAwareness of Patients With Symptomatic Gallstones Regarding Their Own DiseaseHelenCandyPas encore d'évaluation
- Lectura 3seminario Micosis ProfundaDocument8 pagesLectura 3seminario Micosis ProfundaSebastian CastroPas encore d'évaluation
- Renal Disease in Cancer PatientsD'EverandRenal Disease in Cancer PatientsKevin W. FinkelPas encore d'évaluation
- Eslam 2020 Mafld A Consensus Driven Proposed NDocument38 pagesEslam 2020 Mafld A Consensus Driven Proposed NRafael GermanoPas encore d'évaluation
- WameedMUCProject 2022 82426362Document1 pageWameedMUCProject 2022 82426362pgs.imad.sadiqPas encore d'évaluation
- A Premature Neonate LueucicitosisDocument3 pagesA Premature Neonate Lueucicitosisgonococo29Pas encore d'évaluation
- A 38 Year Old WomanDocument12 pagesA 38 Year Old Womandrafq2000Pas encore d'évaluation
- Liver Transplantation - 2007 - Escorsell - Acute Liver Failure in Spain Analysis of 267 CasesDocument7 pagesLiver Transplantation - 2007 - Escorsell - Acute Liver Failure in Spain Analysis of 267 CasesJesús Guzmán DíazPas encore d'évaluation
- A Clinicohaematological Profile of Splenomegaly: Varsha S. Dabadghao, Arundhati G.Diwan, Amol M. RaskarDocument9 pagesA Clinicohaematological Profile of Splenomegaly: Varsha S. Dabadghao, Arundhati G.Diwan, Amol M. RaskarLianSiahaanPas encore d'évaluation
- Belize HistoDocument3 pagesBelize HistoElizabeth DarcyPas encore d'évaluation
- Clinical Presentation of Childhood Leukaemia: A Systematic Review and Meta-AnalysisDocument9 pagesClinical Presentation of Childhood Leukaemia: A Systematic Review and Meta-AnalysisPrima YosiPas encore d'évaluation
- We Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsDocument30 pagesWe Are Intechopen, The World'S Leading Publisher of Open Access Books Built by Scientists, For ScientistsChris NeophytouPas encore d'évaluation
- Emerging Therapeutic Options For Non-Alcoholic Fatty Liver DiseaseDocument13 pagesEmerging Therapeutic Options For Non-Alcoholic Fatty Liver Diseasew57t4yrkt6Pas encore d'évaluation
- Kardon Et Al. - 2000 - Tonsillar Lymphangiomatous Polyps A Clinicopathologic Series of 26 Cases-AnnotatedDocument6 pagesKardon Et Al. - 2000 - Tonsillar Lymphangiomatous Polyps A Clinicopathologic Series of 26 Cases-AnnotatedyoussPas encore d'évaluation
- TiflitisDocument5 pagesTiflitisJosé Luis Navarro RomeroPas encore d'évaluation
- WilsonDocument11 pagesWilsonCarmen OpreaPas encore d'évaluation
- Pi Is 0016508519325685Document14 pagesPi Is 0016508519325685Sefrilia Miftahul JannahPas encore d'évaluation
- Neutropenic Colitis in Children 2011Document4 pagesNeutropenic Colitis in Children 2011IRINA SULEY TIRADO PEREZPas encore d'évaluation
- Pulmonary Impairment After Tuberculosis : Original ResearchDocument8 pagesPulmonary Impairment After Tuberculosis : Original ResearchPalaniappan MeyyappanPas encore d'évaluation
- OK Biblio: Clinical Epidemiology of in Ammatory Bowel DiseaseDocument14 pagesOK Biblio: Clinical Epidemiology of in Ammatory Bowel DiseaseAllan KoschdoskiPas encore d'évaluation
- Endocrinology and Diabetes: Case Studies, Questions and CommentariesD'EverandEndocrinology and Diabetes: Case Studies, Questions and CommentariesPas encore d'évaluation
- BPJ Vol 10 No 3 P 1369-1377Document9 pagesBPJ Vol 10 No 3 P 1369-1377citra annisa fitriPas encore d'évaluation
- Ojped 2024030813565746Document6 pagesOjped 2024030813565746Nicolás Mosso F.Pas encore d'évaluation
- 2018 Article 1600Document4 pages2018 Article 1600christian roblesPas encore d'évaluation
- Lancet Article PDFDocument5 pagesLancet Article PDFBrandy GarciaPas encore d'évaluation
- NIH Public Access: Author ManuscriptDocument12 pagesNIH Public Access: Author ManuscriptadipurnamiPas encore d'évaluation
- Colombia Médica: An Experience of Liver Transplantation in Latin America: A Medical Center in ColombiaDocument6 pagesColombia Médica: An Experience of Liver Transplantation in Latin America: A Medical Center in ColombiaAlejandro Vesga VinchiraPas encore d'évaluation
- Liver Disease: A Clinical CasebookD'EverandLiver Disease: A Clinical CasebookStanley Martin CohenPas encore d'évaluation
- SudaniwawiDocument7 pagesSudaniwawiMouhammed SleiayPas encore d'évaluation
- Upper Tract Urothelial CarcinomaD'EverandUpper Tract Urothelial CarcinomaShahrokh F. ShariatPas encore d'évaluation
- Spinal Tuberculosis in Children: Sarah Eisen, Laura Honywood, Delane Shingadia, Vas NovelliDocument7 pagesSpinal Tuberculosis in Children: Sarah Eisen, Laura Honywood, Delane Shingadia, Vas NovelliAnonymous YaqenULNgPas encore d'évaluation
- Caso Clinico - AdenocarcinomaDocument1 pageCaso Clinico - AdenocarcinomaXana SoaresPas encore d'évaluation
- Recent SeparationDocument6 pagesRecent SeparationEl Karmo SanPas encore d'évaluation
- Progestogen TreatmentDocument6 pagesProgestogen TreatmentIndah HanePas encore d'évaluation
- The Critically Ill Cirrhotic Patient: Evaluation and ManagementD'EverandThe Critically Ill Cirrhotic Patient: Evaluation and ManagementRobert S. RahimiPas encore d'évaluation
- Enfermeda CeliacaDocument22 pagesEnfermeda CeliacaOmar CoroPas encore d'évaluation
- New England Journal Medicine: The ofDocument12 pagesNew England Journal Medicine: The ofannisapritasPas encore d'évaluation
- Management of Wilson Disease: A Pocket GuideD'EverandManagement of Wilson Disease: A Pocket GuideMichael L. SchilskyPas encore d'évaluation
- Gastroesophageal Reflux Disease (Gastroenterology and Hepatology) PDFDocument408 pagesGastroesophageal Reflux Disease (Gastroenterology and Hepatology) PDFaab 1007Pas encore d'évaluation
- Dieta y CancerDocument11 pagesDieta y CancerJose Angel AbadíaPas encore d'évaluation
- Definiendo Fragilidad Desde La GeriatriaDocument10 pagesDefiniendo Fragilidad Desde La GeriatriaYancy Erazo DoradoPas encore d'évaluation
- Final Meta IdfDocument24 pagesFinal Meta IdfdoublejaydPas encore d'évaluation
- Sustained Reduction in The Incidence of Type 2 Diabetes by Lifestyle Intervention: Follow-Up of The Finnish Diabetes Prevention StudyDocument8 pagesSustained Reduction in The Incidence of Type 2 Diabetes by Lifestyle Intervention: Follow-Up of The Finnish Diabetes Prevention StudyYancy Erazo DoradoPas encore d'évaluation
- Use of Glycated Haemoglobin (Hba1C) in The Diagnosis of Diabetes MellitusDocument25 pagesUse of Glycated Haemoglobin (Hba1C) in The Diagnosis of Diabetes MellitusSiskawati Suparmin100% (1)
- Carmela Main Paper EngDocument8 pagesCarmela Main Paper EngRobertoPas encore d'évaluation
- Articles: BackgroundDocument10 pagesArticles: BackgroundYancy Erazo DoradoPas encore d'évaluation
- Journal Medicine: The New EnglandDocument11 pagesJournal Medicine: The New EnglandYancy Erazo DoradoPas encore d'évaluation
- ESC Guidelines On Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration With The EASDDocument63 pagesESC Guidelines On Diabetes, Pre-Diabetes, and Cardiovascular Diseases Developed in Collaboration With The EASDYancy Erazo DoradoPas encore d'évaluation
- Asma y MagnesioDocument5 pagesAsma y MagnesioYancy Erazo DoradoPas encore d'évaluation
- Rabdoide 4Document7 pagesRabdoide 4Yancy Erazo DoradoPas encore d'évaluation
- Standards of Medical Care in Diabetes 2015Document99 pagesStandards of Medical Care in Diabetes 2015Juan Carlos Sánchez Suárez100% (1)
- Rabdoide 1Document5 pagesRabdoide 1Yancy Erazo DoradoPas encore d'évaluation
- Referral To Pediatric Surgical Specialists-Pediatrics 2014Document9 pagesReferral To Pediatric Surgical Specialists-Pediatrics 2014Yancy Erazo DoradoPas encore d'évaluation
- Amenorrea Investigacion y Tratamiento Current 2004Document7 pagesAmenorrea Investigacion y Tratamiento Current 2004Yancy Erazo DoradoPas encore d'évaluation
- BNP Vs NTproBNPDocument15 pagesBNP Vs NTproBNPYancy Erazo DoradoPas encore d'évaluation
- Chloride The Queen of ElectrolytesDocument9 pagesChloride The Queen of ElectrolytesYancy Erazo DoradoPas encore d'évaluation
- Asma y MagnesioDocument5 pagesAsma y MagnesioYancy Erazo DoradoPas encore d'évaluation
- Chronic Abdominal PainDocument4 pagesChronic Abdominal PainYancy Erazo DoradoPas encore d'évaluation
- (SLE) and PregnancyDocument39 pages(SLE) and PregnancyRake SardevaPas encore d'évaluation
- Nursing Care of A Family Experiencing A Postpartal Complications PDFDocument9 pagesNursing Care of A Family Experiencing A Postpartal Complications PDFTandingco, Olivia Mari H.Pas encore d'évaluation
- Numerology 1 - IntroductionDocument15 pagesNumerology 1 - IntroductionReiki Kiran100% (3)
- 2009 Nec Hiv Dec Aidsreg2009Document3 pages2009 Nec Hiv Dec Aidsreg2009Jing CruzPas encore d'évaluation
- ECG and ArrhythmiasDocument25 pagesECG and ArrhythmiasRashed ShatnawiPas encore d'évaluation
- Blood and Tissue NematodesDocument60 pagesBlood and Tissue NematodesDanielle Pecson100% (1)
- Name of Drug Action Indication Contraindication Adverse Reaction Nursing ConsiderationDocument4 pagesName of Drug Action Indication Contraindication Adverse Reaction Nursing ConsiderationClariss AlotaPas encore d'évaluation
- Uterine AtonyDocument4 pagesUterine AtonyThirdie LacortePas encore d'évaluation
- 22 Long Cases in Medicine by Shamol Sir PDFDocument259 pages22 Long Cases in Medicine by Shamol Sir PDFHossain Muhammad80% (5)
- Nasopharyngeal CarcinomaDocument25 pagesNasopharyngeal Carcinomananda surastyo100% (1)
- NCP (Actual)Document2 pagesNCP (Actual)Silinna May Lee SanicoPas encore d'évaluation
- Case Write Up Harmeet Multinodular GoitreDocument29 pagesCase Write Up Harmeet Multinodular GoitreDevikha PeremelPas encore d'évaluation
- Entamoeba Coli: BackgroundDocument4 pagesEntamoeba Coli: BackgroundJam Pelario100% (1)
- Nursing Care Plan Urinary Tract Infection (UTI)Document2 pagesNursing Care Plan Urinary Tract Infection (UTI)deric95% (97)
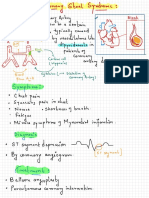
- Coronary Steal SyndromeDocument3 pagesCoronary Steal SyndromeEdward XiamPas encore d'évaluation
- Breast Cancer: Prima Medika HospitalDocument35 pagesBreast Cancer: Prima Medika HospitalPrima MedikaPas encore d'évaluation
- ReflectionDocument4 pagesReflectionelizaviraniPas encore d'évaluation
- Isolated Glomerular Diseases Associated With Recurrent Gross HematuriaDocument17 pagesIsolated Glomerular Diseases Associated With Recurrent Gross HematuriaMay PingolPas encore d'évaluation
- Psychological Problems Associated With Thalassemia in Diyala Province, Iraq - OdtDocument6 pagesPsychological Problems Associated With Thalassemia in Diyala Province, Iraq - OdtThe Swedish Journal of Scientific Research (SJSR) ISSN: 2001-9211Pas encore d'évaluation
- Breast Cancer Awareness - 13 October 2019Document4 pagesBreast Cancer Awareness - 13 October 2019Times MediaPas encore d'évaluation
- Ascites Causes Diagnosis and TreatmentDocument24 pagesAscites Causes Diagnosis and TreatmentrifkaPas encore d'évaluation
- Clobetasol Propionate BP: Scalp SolutionDocument1 pageClobetasol Propionate BP: Scalp SolutionRuhul AminPas encore d'évaluation
- Krok 1 Stomatology 2012Document22 pagesKrok 1 Stomatology 2012Saaha ParmarPas encore d'évaluation
- Apoptosis & Its Relation To CancerDocument16 pagesApoptosis & Its Relation To CancerRohan DuttaPas encore d'évaluation
- State of Lung Cancer 2021Document16 pagesState of Lung Cancer 2021Honolulu Star-AdvertiserPas encore d'évaluation
- Prion Disease: Kelly J. Baldwin, MD Cynthia M. Correll, MDDocument12 pagesPrion Disease: Kelly J. Baldwin, MD Cynthia M. Correll, MDMerari Lugo Ocaña100% (1)
- International Journal of Women's Dermatology: Pallavi Ailawadi, Isha Narang, Vijay K. GargDocument4 pagesInternational Journal of Women's Dermatology: Pallavi Ailawadi, Isha Narang, Vijay K. GargFlamina DerniPas encore d'évaluation
- Serum Lipid Profile As A Predictor of Dengue Severity: A Systematic Review and Meta AnalysisDocument13 pagesSerum Lipid Profile As A Predictor of Dengue Severity: A Systematic Review and Meta Analysisamagno7891Pas encore d'évaluation
- Agrypnia Excitata and Obstructive Apnea in A Patient With Fatal Familial Insomnia From ChinaDocument5 pagesAgrypnia Excitata and Obstructive Apnea in A Patient With Fatal Familial Insomnia From ChinaDayana VieiraPas encore d'évaluation
- W6. Tracheostomy, Introduction To Wound Care & NebulizationDocument4 pagesW6. Tracheostomy, Introduction To Wound Care & NebulizationRHEE ADRIENNE LAI NAVALESPas encore d'évaluation