Vous aimerez peut-être aussi
- Physical Organic Chemistry—Ii: Specially Invited Lectures Presented at the Second IUPAC Conference on Physical Organic Chemistry Held at Noordwijkerhout, Netherlands, 29 April–2 May 1974D'EverandPhysical Organic Chemistry—Ii: Specially Invited Lectures Presented at the Second IUPAC Conference on Physical Organic Chemistry Held at Noordwijkerhout, Netherlands, 29 April–2 May 1974Th. J. De BoerPas encore d'évaluation
- Multistep Synthesis of Hexafluoropropylene: Bonifaz Cristobal (Conway, MA)Document13 pagesMultistep Synthesis of Hexafluoropropylene: Bonifaz Cristobal (Conway, MA)Sp4rkZ87Pas encore d'évaluation
- Full TextDocument256 pagesFull TextkanPas encore d'évaluation
- Aliphatic Compounds: Trihydric Alcohols, Their Oxidation Products and Derivatives, Penta- and Higher Polyhydric Alcohols, Their Oxidation Products and Derivatives; Saccharides, Tetrahydric Alcohols, Their Oxidation Products and DerivativesD'EverandAliphatic Compounds: Trihydric Alcohols, Their Oxidation Products and Derivatives, Penta- and Higher Polyhydric Alcohols, Their Oxidation Products and Derivatives; Saccharides, Tetrahydric Alcohols, Their Oxidation Products and DerivativesPas encore d'évaluation
- United States Patent (19) : Charleston, Both of W. Va.Document10 pagesUnited States Patent (19) : Charleston, Both of W. Va.andari yuta palwaPas encore d'évaluation
- Transition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesD'EverandTransition Metal Catalyzed Furans Synthesis: Transition Metal Catalyzed Heterocycle Synthesis SeriesPas encore d'évaluation
- Hydrates of 1-Methyl-3 - and - 4-Piperidone Hydrochlorides - J Org Chem, 1959, 24 (3), 342 - Jo01085a015Document4 pagesHydrates of 1-Methyl-3 - and - 4-Piperidone Hydrochlorides - J Org Chem, 1959, 24 (3), 342 - Jo01085a015muopioidreceptor100% (1)
- US3891683Document5 pagesUS3891683Risma Dewi SPas encore d'évaluation
- Us ADocument29 pagesUs AamolidPas encore d'évaluation
- United States Patent (19) : 54 Process For Producing AcrylicacidDocument10 pagesUnited States Patent (19) : 54 Process For Producing AcrylicacidKatia Gutierrez GalaPas encore d'évaluation
- Patente Del Ibuprofeno (US4981995)Document16 pagesPatente Del Ibuprofeno (US4981995)María de los Milagros LedesmaPas encore d'évaluation
- Biosynthesis of Valine and Isoleucine: Radhakrishnan, T Wagner, and Esmond SnellDocument10 pagesBiosynthesis of Valine and Isoleucine: Radhakrishnan, T Wagner, and Esmond SnellPatrascu CristinaPas encore d'évaluation
- CR 100258 KDocument35 pagesCR 100258 KzoyudgPas encore d'évaluation
- US6545178Document6 pagesUS6545178Santiago BorgesPas encore d'évaluation
- Us2503724 - Ca2941105a1Document8 pagesUs2503724 - Ca2941105a1Facundo MendezPas encore d'évaluation
- Tetrahydro Quino LinesDocument40 pagesTetrahydro Quino LinesРумен ЛяпчевPas encore d'évaluation
- The NitroparaffinsDocument58 pagesThe NitroparaffinsKybernetikum100% (1)
- Chlorinated Diphenyl OxidesDocument26 pagesChlorinated Diphenyl OxidesBash MatPas encore d'évaluation
- 6 ReportDocument31 pages6 ReportAbhi Butani100% (1)
- Heterocyclization of Barbituric Acid: Synthesis of Novel Condensed PyrimidinesDocument7 pagesHeterocyclization of Barbituric Acid: Synthesis of Novel Condensed PyrimidinesSagar PatilPas encore d'évaluation
- Ilyin 2007Document11 pagesIlyin 2007INFINITY & BEYONDPas encore d'évaluation
- Full Paper: Anna Rosiak, Wolfgang Frey, and Jens ChristoffersDocument11 pagesFull Paper: Anna Rosiak, Wolfgang Frey, and Jens Christofferstakiq lasePas encore d'évaluation
- Synthesis & Characterization of Quinoxalines Chapter-4Document29 pagesSynthesis & Characterization of Quinoxalines Chapter-4Prasada Rao Ch MMPas encore d'évaluation
- 4-Hydroxy Coumarin PatentDocument4 pages4-Hydroxy Coumarin PatentAfzal PathanPas encore d'évaluation
- US PatentDocument4 pagesUS PatentfhafizfrPas encore d'évaluation
- Paten US4223163Document7 pagesPaten US4223163rahmanPas encore d'évaluation
- US6114591Document6 pagesUS6114591Umair NasimPas encore d'évaluation
- Unit-Viii: Synthetic FibersDocument35 pagesUnit-Viii: Synthetic FibersAnilKumarPas encore d'évaluation
- LuzuriagaKenia - PalmaMaría - Post - Practica 4Document20 pagesLuzuriagaKenia - PalmaMaría - Post - Practica 4MARIA DANIELA PALMA LOORPas encore d'évaluation
- 360exp10-02 EsterificationDocument14 pages360exp10-02 EsterificationlewisrahimiPas encore d'évaluation
- Chalcone ThesisDocument29 pagesChalcone ThesisUjjwal Sharma60% (5)
- Current+Organic+Chemistry +2008, 12,+1116-1183Document96 pagesCurrent+Organic+Chemistry +2008, 12,+1116-1183Murali Venkat NagPas encore d'évaluation
- Alkaloids and OthersDocument24 pagesAlkaloids and OtherszakybaaPas encore d'évaluation
- 1 s2.0 0040403996013512 MainDocument4 pages1 s2.0 0040403996013512 MainSupriya SomvanshiPas encore d'évaluation
- US4717720Document12 pagesUS4717720Maria AraujoPas encore d'évaluation
- US4841089Document5 pagesUS4841089Suharti RifaiPas encore d'évaluation
- Piperidine Derivatives. XXI. 4-Piperidone, 4-Piperidinol and Certain of Their Derivatives - J Am Chem Soc, 1949, 71 (3), 901-906 - Ja01171a038Document6 pagesPiperidine Derivatives. XXI. 4-Piperidone, 4-Piperidinol and Certain of Their Derivatives - J Am Chem Soc, 1949, 71 (3), 901-906 - Ja01171a038muopioidreceptorPas encore d'évaluation
- US5349102Document3 pagesUS5349102Tua HalomoanPas encore d'évaluation
- Studies in Piperidone Chemistry. I. A Synthesis of 5-Homopiperazinones - J Org Chem, 1949, 14 (4), 530 - Jo01156a005Document7 pagesStudies in Piperidone Chemistry. I. A Synthesis of 5-Homopiperazinones - J Org Chem, 1949, 14 (4), 530 - Jo01156a005muopioidreceptorPas encore d'évaluation
- Method For Synthesizing PiperonalDocument6 pagesMethod For Synthesizing PiperonalhappylmPas encore d'évaluation
- 4,7-Dichloroquinoline: Molecular Formula: Molecular Weight: Cas NumberDocument5 pages4,7-Dichloroquinoline: Molecular Formula: Molecular Weight: Cas NumberShankar kumar royPas encore d'évaluation
- Intramolecular Acylation of Aryl - and Aroyl-Aliphatic Acids by The Action of Pyrophosphoryl Chloride and Phosphorus OxychlorideDocument8 pagesIntramolecular Acylation of Aryl - and Aroyl-Aliphatic Acids by The Action of Pyrophosphoryl Chloride and Phosphorus OxychlorideChiến NguyễnPas encore d'évaluation
- Exp 10 EsterDocument14 pagesExp 10 EsterLaris J. Garcia100% (1)
- Síntesis de Calix (4) PirrolDocument5 pagesSíntesis de Calix (4) PirrolFelipe De Gante100% (1)
- PDF DocumentDocument33 pagesPDF DocumentNguyễnHuyPhúcPas encore d'évaluation
- Us 2845438Document5 pagesUs 2845438Pat22 22patPas encore d'évaluation
- J. Comb. Chem. 2003, 5, 645-652Document8 pagesJ. Comb. Chem. 2003, 5, 645-652Tsung-Chih ChenPas encore d'évaluation
- ChemSusChem - 2010 - ArceoDocument3 pagesChemSusChem - 2010 - ArceoDanny RonaynePas encore d'évaluation
- Functionalized Tetrahydropyridines by Enantioselective Phosphine-Catalyzed Aza - (4 + 2) Cycloaddition of N Sulfonyl-1-Aza-1,3-Dienes With Vinyl KetonesDocument4 pagesFunctionalized Tetrahydropyridines by Enantioselective Phosphine-Catalyzed Aza - (4 + 2) Cycloaddition of N Sulfonyl-1-Aza-1,3-Dienes With Vinyl KetonesOscar CoralPas encore d'évaluation
- IzatinDocument141 pagesIzatinthamizh555Pas encore d'évaluation
- Synthesis of Stable Isotope Labeled Chloroquine, Amodiaquine and Their MetabolitesDocument27 pagesSynthesis of Stable Isotope Labeled Chloroquine, Amodiaquine and Their MetabolitesthanaPas encore d'évaluation
- US3639461Document5 pagesUS3639461M. IDRISPas encore d'évaluation
- @ Gradeff US3917713 1975 Process For Preparing HydroxycitronellalDocument9 pages@ Gradeff US3917713 1975 Process For Preparing HydroxycitronellalLouisPas encore d'évaluation
- The Highly Selective Oxidation of Cyclohexane To Cyclohexanone and Cyclohexanol Over Valpo Berlinite by Oxygen Under Atmospheric PressureDocument9 pagesThe Highly Selective Oxidation of Cyclohexane To Cyclohexanone and Cyclohexanol Over Valpo Berlinite by Oxygen Under Atmospheric PressureFelipe2 HoyosPas encore d'évaluation
- Venkat Rag A Van 2009Document6 pagesVenkat Rag A Van 2009Irene ValdiviesoPas encore d'évaluation
- UC Berkeley: Green ChemistryDocument4 pagesUC Berkeley: Green ChemistryAndonis AngelovPas encore d'évaluation
- US3903185Document6 pagesUS3903185Muhammad Akbar FahleviPas encore d'évaluation
- MetallurgyDocument39 pagesMetallurgyPrabhakar BandaruPas encore d'évaluation
- Hubungan Tumbuhan Dengan Unsur HaraDocument42 pagesHubungan Tumbuhan Dengan Unsur HaraputriPas encore d'évaluation
- كيمياء حيوية الوحدة التانيةDocument50 pagesكيمياء حيوية الوحدة التانيةasem sawalmehPas encore d'évaluation
- CHM142 Final July 2008Document11 pagesCHM142 Final July 2008Lee_Moi_Yeoh_6863Pas encore d'évaluation
- Fr9-Link1, PL1, FR10-PL5Document4 pagesFr9-Link1, PL1, FR10-PL5Chan ChanPas encore d'évaluation
- Aminoácidos, Petidios e Proteinas Ch23MRDocument52 pagesAminoácidos, Petidios e Proteinas Ch23MRDiogo SantanaPas encore d'évaluation
- Biomolecules PartDocument22 pagesBiomolecules PartYashPas encore d'évaluation
- Physical Chemistry By: Shailendra KumarDocument1 pagePhysical Chemistry By: Shailendra KumarABHIROOP REDDYPas encore d'évaluation
- Chemistry Project : - ADULTERANTS IN FOOD STUFFDocument16 pagesChemistry Project : - ADULTERANTS IN FOOD STUFFArav SinghaniaPas encore d'évaluation
- 10th-C Chapter 12 Re-TestDocument2 pages10th-C Chapter 12 Re-TestWasim NawazPas encore d'évaluation
- Perhitungan Netralisasi Limbah PDFDocument4 pagesPerhitungan Netralisasi Limbah PDFGerry HandoyoPas encore d'évaluation
- Checkpoint Revision Worksheet # 1Document2 pagesCheckpoint Revision Worksheet # 1Sana ShakeelPas encore d'évaluation
- Brochura Lankem - Bio SurfactantesDocument16 pagesBrochura Lankem - Bio SurfactantesleandrolabtecnicoPas encore d'évaluation
- Cargo CompatibilityDocument3 pagesCargo CompatibilityVadim PimenovPas encore d'évaluation
- Acids Bases MCQDocument8 pagesAcids Bases MCQRaahul B SPas encore d'évaluation
- Science Form 2 Chapter 5.4 Solution and Solubility of Substance NoteDocument23 pagesScience Form 2 Chapter 5.4 Solution and Solubility of Substance NoteMiNH Hayat67% (3)
- Pengaruh Konsentrasi Larutan Natrium Klorida (Nacl) Sebagai Bahan Perendam Terhadap Krakteristik Mutu Pati Ubi Talas (Calocasia Esculenta L. Schott)Document11 pagesPengaruh Konsentrasi Larutan Natrium Klorida (Nacl) Sebagai Bahan Perendam Terhadap Krakteristik Mutu Pati Ubi Talas (Calocasia Esculenta L. Schott)No NamePas encore d'évaluation
- Food & Function: ReviewDocument12 pagesFood & Function: ReviewrafacpereiraPas encore d'évaluation
- TitrationDocument4 pagesTitrationYuvrajPas encore d'évaluation
- Certificat Garantie FinalDocument1 pageCertificat Garantie FinalIrina AndreiPas encore d'évaluation
- As 02 Biomolecules Ques - BiologyDocument21 pagesAs 02 Biomolecules Ques - BiologyUncleBulgariaPas encore d'évaluation
- 2 - Lipid BiosynthesisDocument44 pages2 - Lipid BiosynthesisAhmed HamarnehPas encore d'évaluation
- Pat056021-Ep-Etd02 B1Document84 pagesPat056021-Ep-Etd02 B1Elitsa IvanovaPas encore d'évaluation
- Daftar Bahan Kimia PaDocument2 pagesDaftar Bahan Kimia Papanda_oonPas encore d'évaluation
- Research Paper1Document8 pagesResearch Paper1elmonemPas encore d'évaluation
- 0035 0045Document11 pages0035 0045Frank MtetwaPas encore d'évaluation
- MODULE 4 Alkenes and AlkynesDocument19 pagesMODULE 4 Alkenes and AlkynesSittie Fahieda AloyodanPas encore d'évaluation
- Info Ozone Compatible MaterialDocument2 pagesInfo Ozone Compatible MaterialsaracelyPas encore d'évaluation
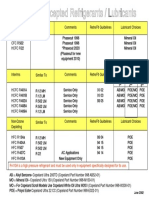
- Copeland Accepted Lubricants 06-28-02Document1 pageCopeland Accepted Lubricants 06-28-02jose antonioPas encore d'évaluation
- Cambridge International Examinations: Chemistry 9701/41 May/June 2017Document12 pagesCambridge International Examinations: Chemistry 9701/41 May/June 2017Robby ReyPas encore d'évaluation
- ChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindD'EverandChatGPT Money Machine 2024 - The Ultimate Chatbot Cheat Sheet to Go From Clueless Noob to Prompt Prodigy Fast! Complete AI Beginner’s Course to Catch the GPT Gold Rush Before It Leaves You BehindPas encore d'évaluation
- The Fabric of Civilization: How Textiles Made the WorldD'EverandThe Fabric of Civilization: How Textiles Made the WorldÉvaluation : 4.5 sur 5 étoiles4.5/5 (58)
- Highest Duty: My Search for What Really MattersD'EverandHighest Duty: My Search for What Really MattersPas encore d'évaluation
- Hero Found: The Greatest POW Escape of the Vietnam WarD'EverandHero Found: The Greatest POW Escape of the Vietnam WarÉvaluation : 4 sur 5 étoiles4/5 (19)
- Sully: The Untold Story Behind the Miracle on the HudsonD'EverandSully: The Untold Story Behind the Miracle on the HudsonÉvaluation : 4 sur 5 étoiles4/5 (103)
- The End of Craving: Recovering the Lost Wisdom of Eating WellD'EverandThe End of Craving: Recovering the Lost Wisdom of Eating WellÉvaluation : 4.5 sur 5 étoiles4.5/5 (81)
- System Error: Where Big Tech Went Wrong and How We Can RebootD'EverandSystem Error: Where Big Tech Went Wrong and How We Can RebootPas encore d'évaluation
- Transformed: Moving to the Product Operating ModelD'EverandTransformed: Moving to the Product Operating ModelÉvaluation : 4 sur 5 étoiles4/5 (1)
- Reality+: Virtual Worlds and the Problems of PhilosophyD'EverandReality+: Virtual Worlds and the Problems of PhilosophyÉvaluation : 4 sur 5 étoiles4/5 (24)
- Faster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestD'EverandFaster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestÉvaluation : 4 sur 5 étoiles4/5 (28)
- Pale Blue Dot: A Vision of the Human Future in SpaceD'EverandPale Blue Dot: A Vision of the Human Future in SpaceÉvaluation : 4.5 sur 5 étoiles4.5/5 (588)
- The Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyD'EverandThe Intel Trinity: How Robert Noyce, Gordon Moore, and Andy Grove Built the World's Most Important CompanyPas encore d'évaluation
- Packing for Mars: The Curious Science of Life in the VoidD'EverandPacking for Mars: The Curious Science of Life in the VoidÉvaluation : 4 sur 5 étoiles4/5 (1395)
- The Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaD'EverandThe Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaPas encore d'évaluation
- How to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerD'EverandHow to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerÉvaluation : 4.5 sur 5 étoiles4.5/5 (54)
- A Place of My Own: The Architecture of DaydreamsD'EverandA Place of My Own: The Architecture of DaydreamsÉvaluation : 4 sur 5 étoiles4/5 (242)
- Fire on the Horizon: The Untold Story of the Gulf Oil DisasterD'EverandFire on the Horizon: The Untold Story of the Gulf Oil DisasterPas encore d'évaluation
- The Future of Geography: How the Competition in Space Will Change Our WorldD'EverandThe Future of Geography: How the Competition in Space Will Change Our WorldÉvaluation : 4 sur 5 étoiles4/5 (5)
- How to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerD'EverandHow to Build a Car: The Autobiography of the World’s Greatest Formula 1 DesignerÉvaluation : 4.5 sur 5 étoiles4.5/5 (122)
- The Weather Machine: A Journey Inside the ForecastD'EverandThe Weather Machine: A Journey Inside the ForecastÉvaluation : 3.5 sur 5 étoiles3.5/5 (31)
- Broken Money: Why Our Financial System is Failing Us and How We Can Make it BetterD'EverandBroken Money: Why Our Financial System is Failing Us and How We Can Make it BetterÉvaluation : 5 sur 5 étoiles5/5 (3)
- The Things We Make: The Unknown History of Invention from Cathedrals to Soda CansD'EverandThe Things We Make: The Unknown History of Invention from Cathedrals to Soda CansPas encore d'évaluation
- Dirt to Soil: One Family’s Journey into Regenerative AgricultureD'EverandDirt to Soil: One Family’s Journey into Regenerative AgricultureÉvaluation : 5 sur 5 étoiles5/5 (125)
- The Technology Trap: Capital, Labor, and Power in the Age of AutomationD'EverandThe Technology Trap: Capital, Labor, and Power in the Age of AutomationÉvaluation : 4.5 sur 5 étoiles4.5/5 (46)
- Data-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseD'EverandData-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseÉvaluation : 3.5 sur 5 étoiles3.5/5 (12)