Vous aimerez peut-être aussi
- Kindergarten Summer Homework PacketDocument36 pagesKindergarten Summer Homework PacketSeravina JovithaPas encore d'évaluation
- PyLammps TutorialDocument15 pagesPyLammps TutorialmaludeavPas encore d'évaluation
- The Elements of C++ Style PDFDocument188 pagesThe Elements of C++ Style PDFfikretgulsoyPas encore d'évaluation
- EDCDocument67 pagesEDCKiran VeesamPas encore d'évaluation
- The User's Guide To The Solar Dynamo Code SuryaDocument16 pagesThe User's Guide To The Solar Dynamo Code Suryasoumitrahazra100% (1)
- Hetro JunctionDocument30 pagesHetro JunctionKamalesh DebnathPas encore d'évaluation
- Project ReportDocument24 pagesProject ReportBahroz RashidPas encore d'évaluation
- Shankar Exercises 05.01.01Document3 pagesShankar Exercises 05.01.01Priyaranjan SahooPas encore d'évaluation
- LAMMPS TutorialDocument96 pagesLAMMPS TutorialNABIL HUSSAINPas encore d'évaluation
- LAMMPS For BeginnersDocument41 pagesLAMMPS For Beginnersaravindrammohan100% (3)
- Challenges For Density Functional TheoryDocument32 pagesChallenges For Density Functional TheoryLucas FagundesPas encore d'évaluation
- Quantum Espresso Inputfile ExplantionDocument31 pagesQuantum Espresso Inputfile ExplantionPHYSICS PROBLEMSPas encore d'évaluation
- BigDFT Manual 1.4Document35 pagesBigDFT Manual 1.4Sunghyun KimPas encore d'évaluation
- LAMMPSDocument92 pagesLAMMPSdaniyalhassan2789Pas encore d'évaluation
- Toolbox DFTDocument20 pagesToolbox DFTAshok Kumar JhaPas encore d'évaluation
- How To Execute Materials StudioDocument7 pagesHow To Execute Materials StudioHirak ChatterjeePas encore d'évaluation
- Atomic Scale Calculations of Tungsten Surface Binding Energy and Be Induced Tungsten SputteringDocument4 pagesAtomic Scale Calculations of Tungsten Surface Binding Energy and Be Induced Tungsten Sputteringimdad_buetPas encore d'évaluation
- Introduction To Density Functional TheoryDocument44 pagesIntroduction To Density Functional TheoryredaelwanPas encore d'évaluation
- MgO VASP TutorialDocument16 pagesMgO VASP TutorialKartavya BholaPas encore d'évaluation
- Structure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsD'EverandStructure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsPas encore d'évaluation
- DFT Using PythonDocument286 pagesDFT Using PythonAvanindra Kumar Pandeya100% (5)
- Tight Density Functional Theory PHD ThesisDocument98 pagesTight Density Functional Theory PHD ThesisNg Wei JiangPas encore d'évaluation
- Vasp Lab1 IntroDocument6 pagesVasp Lab1 IntroSam RoelantsPas encore d'évaluation
- Multiscale Simulation Methods PDFDocument592 pagesMultiscale Simulation Methods PDFFredrick Antony0% (1)
- DFT MetaisDocument60 pagesDFT MetaisGiliandroFariasPas encore d'évaluation
- Quantum ExpressoDocument49 pagesQuantum ExpressoDavidAlejoPas encore d'évaluation
- Figure of Merit PHOTO DIODEDocument26 pagesFigure of Merit PHOTO DIODEAnonymous eWMnRr70qPas encore d'évaluation
- Atomic Force MicrosDocument42 pagesAtomic Force MicrosC. GaussPas encore d'évaluation
- Praktikum 4 18122014Document31 pagesPraktikum 4 18122014George Michael Alvarado LopezPas encore d'évaluation
- Density Functional TheoryDocument9 pagesDensity Functional TheoryVarovPas encore d'évaluation
- Surface ChemistryDocument137 pagesSurface ChemistryMohammed86% (7)
- Tight BindingDocument5 pagesTight BindingalkeronePas encore d'évaluation
- VAPS Tutorial11 01 2010Document38 pagesVAPS Tutorial11 01 2010Clarence AG Yue100% (1)
- Quantum MechanicsDocument11 pagesQuantum MechanicsPrajan PoudelPas encore d'évaluation
- 4-Lesson4-Intrinsic & Doped Semiconductor (Compatibility Mode)Document26 pages4-Lesson4-Intrinsic & Doped Semiconductor (Compatibility Mode)reza oemarPas encore d'évaluation
- Chapter11 PDFDocument8 pagesChapter11 PDFpapipoPas encore d'évaluation
- First Step To Computer Programming With FORTRAN 90Document14 pagesFirst Step To Computer Programming With FORTRAN 90Omed Ghareb100% (2)
- Surface Enhanced Raman SpectrosDocument23 pagesSurface Enhanced Raman SpectrosRevati MutturPas encore d'évaluation
- Designing Analog ChipsDocument242 pagesDesigning Analog Chipsapi-3746617Pas encore d'évaluation
- Miura - Physics of Semiconductors in High Magnetic FieldsDocument373 pagesMiura - Physics of Semiconductors in High Magnetic Fieldsaljegu1Pas encore d'évaluation
- 2.2 Elementary Introduction To Molecular Mechanics and DynamicsDocument13 pages2.2 Elementary Introduction To Molecular Mechanics and Dynamicsanu_prabhaPas encore d'évaluation
- Quantum Espresso TutorialDocument23 pagesQuantum Espresso TutorialCarla Vieira SoaresPas encore d'évaluation
- 10.1201 9781003290964 PreviewpdfDocument20 pages10.1201 9781003290964 PreviewpdfmokhtarkanPas encore d'évaluation
- Computational ChemistryDocument10 pagesComputational ChemistryRenjith Raveendran Pillai100% (1)
- ComputationalDocument300 pagesComputationalAli GaGo100% (1)
- Atomic and Molecular Structure NotesDocument20 pagesAtomic and Molecular Structure Noteske.Pas encore d'évaluation
- Heterostructurefundamentals PDFDocument43 pagesHeterostructurefundamentals PDFADITYA SINGHPas encore d'évaluation
- Gaussian TutorialDocument36 pagesGaussian TutorialandreaPas encore d'évaluation
- DFT in PracticeDocument33 pagesDFT in PracticeChang Jae LeePas encore d'évaluation
- Advanced Quantum Mehanics-Peter S. RiseboroughDocument379 pagesAdvanced Quantum Mehanics-Peter S. Riseboroughsilv_erPas encore d'évaluation
- Foundations of Quantum MechanicsDocument35 pagesFoundations of Quantum MechanicsYi ChaiPas encore d'évaluation
- Memristor PPT FinalDocument23 pagesMemristor PPT FinalIndraprakas KnPas encore d'évaluation
- Free and Open Source Software For Computational Chemistry EducationDocument30 pagesFree and Open Source Software For Computational Chemistry EducationFernando CisnerosPas encore d'évaluation
- Superconductivity PresentationDocument26 pagesSuperconductivity PresentationSabrina Palazzese Di BasilioPas encore d'évaluation
- De Haas Van Alphen EffectDocument13 pagesDe Haas Van Alphen EffectbillcosbyfatherhoodPas encore d'évaluation
- PDFDocument7 pagesPDFSayantan0% (2)
- IntroMDsimulations WGwebinar 01nov2017Document76 pagesIntroMDsimulations WGwebinar 01nov2017Rizal SinagaPas encore d'évaluation
- Atomic and Molecular Physics - NotesDocument200 pagesAtomic and Molecular Physics - NotesAasim PatelPas encore d'évaluation
- STM TheoryDocument38 pagesSTM TheoryAmintujy Singh RajputPas encore d'évaluation
- Grep Awk SedDocument9 pagesGrep Awk SedvishnuselvaPas encore d'évaluation
- Modeling of C NanotubesDocument10 pagesModeling of C NanotubesMuraleetharan BoopathiPas encore d'évaluation
- NBODocument8 pagesNBOA Fernando CGPas encore d'évaluation
- New Frontiers in Graph TheoryDocument526 pagesNew Frontiers in Graph TheorySheshera Mysore100% (1)
- An Introduction To Python ProgrammingDocument51 pagesAn Introduction To Python ProgrammingNgocLienHoaNguyenPas encore d'évaluation
- Dimeros Ácido FórmicoDocument10 pagesDimeros Ácido FórmicoA Fernando CGPas encore d'évaluation
- CARL J. WENNING - Scientific Epistemology - How Scientists Know What They Know (JPTEO - Autumn 2009)Document13 pagesCARL J. WENNING - Scientific Epistemology - How Scientists Know What They Know (JPTEO - Autumn 2009)A Fernando CGPas encore d'évaluation
- Top 500 English To GermanDocument22 pagesTop 500 English To Germangerman vocabularyPas encore d'évaluation
- HOW Singles Advanced Typography On The WebDocument4 pagesHOW Singles Advanced Typography On The Webjuliorivas2136Pas encore d'évaluation
- 'Political Animals' and The Sinhala Thuppahi ManDocument54 pages'Political Animals' and The Sinhala Thuppahi ManRathnapala SubasinghePas encore d'évaluation
- Traditions and Encounters A Global Perspective On The Past 6th Edition Bentley Test Bank PDFDocument15 pagesTraditions and Encounters A Global Perspective On The Past 6th Edition Bentley Test Bank PDFa501579707Pas encore d'évaluation
- 10 Grade Leadership Research Paper Rubric NameDocument2 pages10 Grade Leadership Research Paper Rubric Name2theMoon_js8242Pas encore d'évaluation
- French German Spanish 2019 20Document59 pagesFrench German Spanish 2019 20cliffordjose2001Pas encore d'évaluation
- Maududi BiographyDocument14 pagesMaududi BiographyRehmat Ali0% (1)
- Action Research Proposal DoneDocument19 pagesAction Research Proposal DoneMuhammad Fauzul Fikri100% (13)
- The Comedy of Errors Thesis StatementDocument4 pagesThe Comedy of Errors Thesis Statementtracyrichardsomaha100% (2)
- English Language (English Paper - 1) PDFDocument7 pagesEnglish Language (English Paper - 1) PDFAyana DeyPas encore d'évaluation
- 1882 - B. Jülg - On The Present State of Mongolian ResearchesDocument24 pages1882 - B. Jülg - On The Present State of Mongolian Researchesfatih çiftçiPas encore d'évaluation
- Nonlinear PhonologyDocument26 pagesNonlinear PhonologyCaro ContrerasPas encore d'évaluation
- Demonstrativo - NewFast English - Book3Document16 pagesDemonstrativo - NewFast English - Book3Maicon BritoPas encore d'évaluation
- The Role of The First LanguageDocument1 pageThe Role of The First Languagequynhthy189Pas encore d'évaluation
- The Transformation of Architectural Narrative From Literature To Cinema PDFDocument129 pagesThe Transformation of Architectural Narrative From Literature To Cinema PDFSelcen YeniçeriPas encore d'évaluation
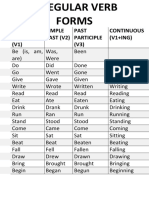
- Irregular Verb Forms (Potrait)Document4 pagesIrregular Verb Forms (Potrait)MaruthiPas encore d'évaluation
- Bai Giang Chi Tiet Tieng Anh 11Document277 pagesBai Giang Chi Tiet Tieng Anh 11tuantu_5160% (5)
- Soal Bahasa Inggris USM STIS 2013: Waktu: 60 Menit PetunjukDocument9 pagesSoal Bahasa Inggris USM STIS 2013: Waktu: 60 Menit PetunjukDaniel AprianusPas encore d'évaluation
- Tu Get2540Document60 pagesTu Get2540nawapatPas encore d'évaluation
- Stephen Krashen's Theory of Second Language AcquisitionDocument5 pagesStephen Krashen's Theory of Second Language AcquisitionNandadewa Aji PutraPas encore d'évaluation
- Newsletters 2006 - AVTDocument32 pagesNewsletters 2006 - AVTAna VellegalPas encore d'évaluation
- Information Distribution Aspects of Design MethodologyDocument27 pagesInformation Distribution Aspects of Design MethodologyAmir NaghizadehPas encore d'évaluation
- Brenna Bhandar, Jonathan Goldberg-Hiller - Plastic Materialities - Politics, Legality, and Metamorphosis in The Work of Catherine Malabou (2015, Duke University Press Books) PDFDocument348 pagesBrenna Bhandar, Jonathan Goldberg-Hiller - Plastic Materialities - Politics, Legality, and Metamorphosis in The Work of Catherine Malabou (2015, Duke University Press Books) PDFiamjacksbPas encore d'évaluation
- Rizal's Secret MissionDocument3 pagesRizal's Secret MissionBadet ReyesPas encore d'évaluation
- People Also Ask: A - WikipediaDocument5 pagesPeople Also Ask: A - WikipediaE-zat IlmanPas encore d'évaluation
- Ficha Semanal Del Proyecto Disciplinar II Week 4 10thDocument7 pagesFicha Semanal Del Proyecto Disciplinar II Week 4 10thJordan ToaquizaPas encore d'évaluation
- Ritchie Fabulae FacilesDocument140 pagesRitchie Fabulae Facilesrbruma100% (1)
- Verb TensesDocument35 pagesVerb TensesTiê FelixPas encore d'évaluation
- 1Document3 pages1Sib Sankar RoyPas encore d'évaluation