Vous aimerez peut-être aussi
- Next Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsD'EverandNext Generation Sequencing and Sequence Assembly: Methodologies and AlgorithmsPas encore d'évaluation
- DNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensD'EverandDNA Methods in Food Safety: Molecular Typing of Foodborne and Waterborne Bacterial PathogensPas encore d'évaluation
- Poulain2017NanoCAGE A Method For The Analysis of Coding and Noncoding 5'-Capped TranscriptomesDocument53 pagesPoulain2017NanoCAGE A Method For The Analysis of Coding and Noncoding 5'-Capped TranscriptomesKito TongHuiPas encore d'évaluation
- Takahashi, 2012 5' End-Centered Expression Profiling Using Cap-Analysis Gene Expression and Next-Generation SequencingDocument43 pagesTakahashi, 2012 5' End-Centered Expression Profiling Using Cap-Analysis Gene Expression and Next-Generation SequencingKito TongHuiPas encore d'évaluation
- Perspectives: Rna-Seq: A Revolutionary Tool For TranscriptomicsDocument7 pagesPerspectives: Rna-Seq: A Revolutionary Tool For Transcriptomicsjubatus.libroPas encore d'évaluation
- News and Views: Advancing RNA-Seq AnalysisDocument3 pagesNews and Views: Advancing RNA-Seq AnalysisWilson Rodrigo Cruz FlorPas encore d'évaluation
- RNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsDocument6 pagesRNA Sequencing: An Introduction To Efficient Planning and Execution of RNA Sequencing (RNA-Seq) ExperimentsnareshPas encore d'évaluation
- KB FileDocument18 pagesKB FileKaushik BanikPas encore d'évaluation
- Several Methods For Quantitative Analysis of The TranscriptomeDocument12 pagesSeveral Methods For Quantitative Analysis of The TranscriptomeSeeni BalajiPas encore d'évaluation
- Expression ProfilingDocument8 pagesExpression ProfilingSreedurgalakshmi KPas encore d'évaluation
- Gene PredictionDocument15 pagesGene PredictionRaghav SureshPas encore d'évaluation
- More Translation 2020 Exam Questions and AnswersDocument9 pagesMore Translation 2020 Exam Questions and Answersryan gillPas encore d'évaluation
- Next Generation SequencingDocument70 pagesNext Generation SequencingARUN KUMARPas encore d'évaluation
- Question 5. Mol Base Gene Expression Exam 2021Document2 pagesQuestion 5. Mol Base Gene Expression Exam 2021varasamaliaPas encore d'évaluation
- Lab Report Format Exp 5 Detection of Open Reading Frame by ORF FinderDocument8 pagesLab Report Format Exp 5 Detection of Open Reading Frame by ORF FinderPrasenjit NaskarPas encore d'évaluation
- Analysis of The Wild-Type: C. Elegans TranscriptomeDocument27 pagesAnalysis of The Wild-Type: C. Elegans TranscriptomeFeseha AbebePas encore d'évaluation
- Para, Gene DisDocument11 pagesPara, Gene DisMudit MisraPas encore d'évaluation
- Race PCR DissertationDocument8 pagesRace PCR DissertationWriteMyPapersDiscountCodeUK100% (1)
- BTC 506 Gene Identification Using Bioinformatic Tools-230302130331Document14 pagesBTC 506 Gene Identification Using Bioinformatic Tools-230302130331chijioke NsoforPas encore d'évaluation
- Cloning StrategiesDocument35 pagesCloning Strategiesskmishra8467% (3)
- GPB Notes part-IIDocument37 pagesGPB Notes part-IIChannabasavaPas encore d'évaluation
- Sage Technology and Its ApplicationsDocument61 pagesSage Technology and Its Applicationsthamizh555Pas encore d'évaluation
- Amplified Fragment Length Polymorphism (Aflp)Document36 pagesAmplified Fragment Length Polymorphism (Aflp)Muhammad Achin YasinPas encore d'évaluation
- Presentation On: Gene Expression Analysis - StsDocument14 pagesPresentation On: Gene Expression Analysis - StsIda Nancy KPas encore d'évaluation
- 2016-RNAseq-The Impact of Amplification On Diferential Expresion Analyses by RNA-seqDocument11 pages2016-RNAseq-The Impact of Amplification On Diferential Expresion Analyses by RNA-seqAntarToumaPas encore d'évaluation
- Continuous Assessment: Concentration's Increment in Case of Insert and LigaseDocument8 pagesContinuous Assessment: Concentration's Increment in Case of Insert and LigaseDebapriya HazraPas encore d'évaluation
- Development of A Rapid and Inexpensive Method To Reveal Natural Antisense TranscriptsDocument8 pagesDevelopment of A Rapid and Inexpensive Method To Reveal Natural Antisense Transcriptschandran.vkrsdPas encore d'évaluation
- Overview of Single Molecule Long RNA Seq Reads and Their Transcriptomic Applications in Plant ResearchDocument3 pagesOverview of Single Molecule Long RNA Seq Reads and Their Transcriptomic Applications in Plant ResearchPaul HartingPas encore d'évaluation
- Correct BaseDocument2 pagesCorrect BaseRobotrixPas encore d'évaluation
- Construction of Genomic and cDNA Libraries-AmitDocument10 pagesConstruction of Genomic and cDNA Libraries-AmitPrashant BajpaiPas encore d'évaluation
- Methods To Study Gene LocationdocxDocument2 pagesMethods To Study Gene LocationdocxRanu AgarwalPas encore d'évaluation
- Gene PredictionDocument36 pagesGene PredictionAtul Kumar25% (4)
- Cross-Mapping MicroRNA IdentificationDocument8 pagesCross-Mapping MicroRNA IdentificationRicardo GorePas encore d'évaluation
- BGi RNA-Seq AnalysisDocument19 pagesBGi RNA-Seq Analysisshashikanth_marriPas encore d'évaluation
- Lecture 4: Detecting Mutations and Mutation Consequences Learning GoalsDocument11 pagesLecture 4: Detecting Mutations and Mutation Consequences Learning GoalsAngelica SmithPas encore d'évaluation
- Selecting Open Reading Frames From DNADocument12 pagesSelecting Open Reading Frames From DNAFede JazzPas encore d'évaluation
- SAGE Is Sequence Sampling Technique in Which Very Short Sequence Tags Are Joined Into Long ConcatamersDocument1 pageSAGE Is Sequence Sampling Technique in Which Very Short Sequence Tags Are Joined Into Long ConcatamersSoumitra NathPas encore d'évaluation
- B4 Gene ArraysDocument15 pagesB4 Gene ArraysRizkia Milladina HidayatullohPas encore d'évaluation
- Ponting, Et Al 2009. Evolution and Functions of Long Noncoding RNAs.Document13 pagesPonting, Et Al 2009. Evolution and Functions of Long Noncoding RNAs.J Alberto LucasPas encore d'évaluation
- Rna Seq DissertationDocument6 pagesRna Seq DissertationCanIPaySomeoneToWriteMyPaperSingapore100% (1)
- CH 09 PDFDocument17 pagesCH 09 PDFSyed Ali Akbar BokhariPas encore d'évaluation
- Molecular Markers and Their Characteristics M. R. Meena, M.L. Chhabra, Ravinder Kumar and C. M. MeenaDocument5 pagesMolecular Markers and Their Characteristics M. R. Meena, M.L. Chhabra, Ravinder Kumar and C. M. Meenamintu_uasPas encore d'évaluation
- Method and Result:: 2.1. Sg-RNA DesigningDocument9 pagesMethod and Result:: 2.1. Sg-RNA DesigningMasum Billah TuhinPas encore d'évaluation
- Genome AnnotationDocument58 pagesGenome Annotationronnny11Pas encore d'évaluation
- Rnaseq and Chip-Seq Principles: A) Quantifying Against A GenomeDocument7 pagesRnaseq and Chip-Seq Principles: A) Quantifying Against A Genomehesham12345Pas encore d'évaluation
- Kon Erm Ann 2014Document18 pagesKon Erm Ann 2014Letícia AlibertiPas encore d'évaluation
- BIO353 Lecture 10 mRNA SplicingDocument8 pagesBIO353 Lecture 10 mRNA SplicingMina KoçPas encore d'évaluation
- Next Generation Sequencing (NGS) - FAQSDocument9 pagesNext Generation Sequencing (NGS) - FAQShesham12345Pas encore d'évaluation
- Sequencing 16S rRNA Gene Fragments Using The PacBiDocument26 pagesSequencing 16S rRNA Gene Fragments Using The PacBiSuran Ravindran NambisanPas encore d'évaluation
- Molecular Markers: Types of Genetic MarkersDocument9 pagesMolecular Markers: Types of Genetic Markerssunayana debPas encore d'évaluation
- Group # 13Document49 pagesGroup # 13maheen fatimaPas encore d'évaluation
- Trapnell 2009Document7 pagesTrapnell 2009László SágiPas encore d'évaluation
- Functional GenomicsDocument11 pagesFunctional Genomicswatson191Pas encore d'évaluation
- Gene ChipsDocument12 pagesGene ChipsRabia KhattakPas encore d'évaluation
- International Journal of Computational Engineering Research (IJCER)Document5 pagesInternational Journal of Computational Engineering Research (IJCER)International Journal of computational Engineering research (IJCER)Pas encore d'évaluation
- Art ResDocument6 pagesArt ResAyeshah RosdahPas encore d'évaluation
- Aflp-Pcr:: Steps: 1.DNA ExtractionDocument3 pagesAflp-Pcr:: Steps: 1.DNA ExtractionArsalan Abbas ZafarPas encore d'évaluation
- Lab Assignment 371Document9 pagesLab Assignment 371khanyiso kalyataPas encore d'évaluation
- 1.2,3 DNA SequencingDocument64 pages1.2,3 DNA SequencingMansiPas encore d'évaluation
- Eleven Golden Rules of RT PCRDocument2 pagesEleven Golden Rules of RT PCRDr-Muhammad Atif AttariPas encore d'évaluation
- Optical Fiber Communication 06EC7 2: Citstudents - inDocument4 pagesOptical Fiber Communication 06EC7 2: Citstudents - inShailaja UdtewarPas encore d'évaluation
- Surface Finish StandardDocument3 pagesSurface Finish StandardvinodmysorePas encore d'évaluation
- The Mpeg Dash StandardDocument6 pagesThe Mpeg Dash Standard9716755397Pas encore d'évaluation
- Indicsoft ProfileDocument13 pagesIndicsoft ProfileMahmood AkhtarPas encore d'évaluation
- 1169a AopDocument12 pages1169a AopBAYARD BernardPas encore d'évaluation
- Mals-11, Family Readiness Newsletter, July 2011, The Devilfish TidesDocument9 pagesMals-11, Family Readiness Newsletter, July 2011, The Devilfish TidesDevilfish FROPas encore d'évaluation
- Ic 1403 2 MarksDocument14 pagesIc 1403 2 MarksJeya Prakash100% (2)
- ADP 1 LAB ManualDocument54 pagesADP 1 LAB ManualdhanajayanPas encore d'évaluation
- Wavin QuickStream Specification Oct 14Document6 pagesWavin QuickStream Specification Oct 14Syahrisal SaputraPas encore d'évaluation
- Government Polytechnic Muzaffarpur: Name of The Lab: Power Electronics & DrivesDocument18 pagesGovernment Polytechnic Muzaffarpur: Name of The Lab: Power Electronics & DrivesVK DPas encore d'évaluation
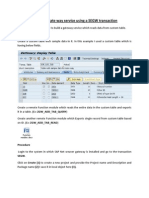
- Building A Gate Way Service Using A SEGW TransactionDocument10 pagesBuilding A Gate Way Service Using A SEGW Transactionswamy_yalamanchiliPas encore d'évaluation
- Sample Problem #8Document8 pagesSample Problem #8DozdiPas encore d'évaluation
- Manual New AswanDocument18 pagesManual New AswanmohamedmosallamPas encore d'évaluation
- Appendix 1: Lesson Plan (Template)Document4 pagesAppendix 1: Lesson Plan (Template)Anonymous X7u8sMPas encore d'évaluation
- Pawan Kumar Dubey: ProfileDocument4 pagesPawan Kumar Dubey: Profilepawandubey9Pas encore d'évaluation
- COOLANT Komatsu PDFDocument16 pagesCOOLANT Komatsu PDFdarwin100% (1)
- Inlet Modules Model Code: ExampleDocument1 pageInlet Modules Model Code: ExamplemhasansharifiPas encore d'évaluation
- Specification For Foamed Concrete: by K C Brady, G R A Watts and Ivi R JonesDocument20 pagesSpecification For Foamed Concrete: by K C Brady, G R A Watts and Ivi R JonesMaria Aiza Maniwang Calumba100% (1)
- SB658Document3 pagesSB658ricardo novoa saraviaPas encore d'évaluation
- Time Impact Analysis by BrewerConsultingDocument3 pagesTime Impact Analysis by BrewerConsultingAAKPas encore d'évaluation
- Swiss FCM Ordinance - EuPIA QDocument6 pagesSwiss FCM Ordinance - EuPIA Qiz_nazPas encore d'évaluation
- Netbackup UsefulDocument56 pagesNetbackup Usefuldnyan123Pas encore d'évaluation
- Pre Joining Formalities Campus Medical FormDocument18 pagesPre Joining Formalities Campus Medical Formrafii_babu1988Pas encore d'évaluation
- Ford Fiesta Specifications: Exterior DimensionsDocument2 pagesFord Fiesta Specifications: Exterior DimensionsBuhuntuUhuPas encore d'évaluation
- Growth of Escherichia Coli in A 5 Litre Batch Fermentation VesselDocument15 pagesGrowth of Escherichia Coli in A 5 Litre Batch Fermentation VesselAs'ad Mughal100% (2)
- SP2000 COD Testkits 2014Document5 pagesSP2000 COD Testkits 2014Andres FalmacelPas encore d'évaluation
- Model Building Intermediate: Exercise GuideDocument64 pagesModel Building Intermediate: Exercise GuideDragos ZvincuPas encore d'évaluation
- Fastcap 22 630v PDFDocument1 pageFastcap 22 630v PDFDarrenPas encore d'évaluation
- Catalog07 PDFDocument13 pagesCatalog07 PDFdeig15050% (2)
- PowerMaxExpress V17 Eng User Guide D-303974 PDFDocument38 pagesPowerMaxExpress V17 Eng User Guide D-303974 PDFSretenPas encore d'évaluation