Vous aimerez peut-être aussi
- Charcot Marie Tooth DiseaseDocument4 pagesCharcot Marie Tooth DiseaseAbdul QuyyumPas encore d'évaluation
- Charcot-Marie-Tooth Disease and Other Hereditary NeuropathiesDocument33 pagesCharcot-Marie-Tooth Disease and Other Hereditary NeuropathiesTallulah Spina TensiniPas encore d'évaluation
- Health and Safety Fact Sheets On Cell TowersDocument19 pagesHealth and Safety Fact Sheets On Cell TowersJoel MilinskyPas encore d'évaluation
- Charcot - MarieDocument4 pagesCharcot - MarieAlexandra MardarePas encore d'évaluation
- HFN 9 - b12 CobalaminDocument5 pagesHFN 9 - b12 CobalaminOvigorPas encore d'évaluation
- NIH Toolbox App Administrator's Manual v1.23 PDFDocument279 pagesNIH Toolbox App Administrator's Manual v1.23 PDFThe English TeacherPas encore d'évaluation
- 2009 QEEG For TBI KThorntonDocument10 pages2009 QEEG For TBI KThorntonmsensoriumPas encore d'évaluation
- Anhedonia As A Central Factor in Depression)Document18 pagesAnhedonia As A Central Factor in Depression)Gkani ChrysoulaPas encore d'évaluation
- Declaration of Doctors Masel and O'Shanick in Support of Brain Injury Association of America Motion For Leave To Be Amicus Curiae in NFL Concussion LitigationDocument11 pagesDeclaration of Doctors Masel and O'Shanick in Support of Brain Injury Association of America Motion For Leave To Be Amicus Curiae in NFL Concussion LitigationGeorge ConkPas encore d'évaluation
- Vitamin b12 Production - ParulDocument20 pagesVitamin b12 Production - Parulanon-90455980% (5)
- 3433 Esketamine Market Shaping Guidebook Final V - 3.5Document230 pages3433 Esketamine Market Shaping Guidebook Final V - 3.5Phương NhungPas encore d'évaluation
- Principles of Tinnitology Tinnitus Diagnosis and Treatment A Tinnitustargeted Therapy PDFDocument13 pagesPrinciples of Tinnitology Tinnitus Diagnosis and Treatment A Tinnitustargeted Therapy PDFbikemanPas encore d'évaluation
- Reading Facial Expressions of EmotionDocument11 pagesReading Facial Expressions of Emotionlinux101053Pas encore d'évaluation
- The Anti Reward Brain SystemDocument42 pagesThe Anti Reward Brain Systemtadcp100% (1)
- CCJM Essential Tremor Choosing The Right Management Plan For Your PatientDocument8 pagesCCJM Essential Tremor Choosing The Right Management Plan For Your PatientBrian HarrisPas encore d'évaluation
- CTE StudyDocument7 pagesCTE StudyTom LeyPas encore d'évaluation
- ICD-11 RevisionDocument22 pagesICD-11 RevisiondfriadyPas encore d'évaluation
- Active Shooter Incidents 2000-2017Document29 pagesActive Shooter Incidents 2000-2017Stephen LoiaconiPas encore d'évaluation
- Testing The Glutamate Hypothesis of SchizophreniaDocument3 pagesTesting The Glutamate Hypothesis of SchizophreniaJohn000123Pas encore d'évaluation
- Lecture Neurotransmitters in CNS and PNSDocument21 pagesLecture Neurotransmitters in CNS and PNSTeePas encore d'évaluation
- 09 Central Nervous System Coloring BookDocument11 pages09 Central Nervous System Coloring BookArmy JmPas encore d'évaluation
- Biological Effects of Electromagnetic Radiation: 19th CenturyDocument23 pagesBiological Effects of Electromagnetic Radiation: 19th CenturydimlevsPas encore d'évaluation
- Omar Amin Final Testimonmial ArticleDocument8 pagesOmar Amin Final Testimonmial ArticleFrancesca HamiltonPas encore d'évaluation
- Spikevax Previously Covid 19 Vaccine Moderna Epar Public Assessment Report - enDocument169 pagesSpikevax Previously Covid 19 Vaccine Moderna Epar Public Assessment Report - enBlessworkPas encore d'évaluation
- Connection of Gallstones To Memory ProblemsDocument7 pagesConnection of Gallstones To Memory ProblemsJacobPadillaPas encore d'évaluation
- Neurostimulation For Stroke RehabilitationDocument11 pagesNeurostimulation For Stroke Rehabilitationisos.mporei.vevaios.Pas encore d'évaluation
- SentrySuite-Software BR EN PDFDocument9 pagesSentrySuite-Software BR EN PDFLajoskaPas encore d'évaluation
- Charcot Marie Tooth - DiseaseDocument11 pagesCharcot Marie Tooth - Diseasenevelle4667Pas encore d'évaluation
- Transversemyelitis: Shin C. Beh,, Benjamin M. Greenberg,, Teresa Frohman,, Elliot M. FrohmanDocument60 pagesTransversemyelitis: Shin C. Beh,, Benjamin M. Greenberg,, Teresa Frohman,, Elliot M. FrohmanRoxana CioflîncPas encore d'évaluation
- Charcot - Marie-Tooth Disease: Signs and SymptomsDocument10 pagesCharcot - Marie-Tooth Disease: Signs and SymptomsIlyas Khan AurakzaiPas encore d'évaluation
- Hirayama's DiseaseDocument27 pagesHirayama's DiseaseDarshika Vyas MohanPas encore d'évaluation
- Peripheral NeuropathyDocument4 pagesPeripheral NeuropathyValerrie NgenoPas encore d'évaluation
- Rare Disease DatabaseDocument8 pagesRare Disease DatabaseBlueash BehPas encore d'évaluation
- Neuromuscular DisordersDocument12 pagesNeuromuscular DisordersZed HarrisPas encore d'évaluation
- 2022 Masingue P.Latour Rev Neurol Genetic Analysis in HereditaryDocument20 pages2022 Masingue P.Latour Rev Neurol Genetic Analysis in HereditaryLéo VidoniPas encore d'évaluation
- Charcot-Marie-Tooth DiseaseDocument9 pagesCharcot-Marie-Tooth Diseaseilyas9558Pas encore d'évaluation
- Kelainan Pada Sistem Saraf PeriferDocument144 pagesKelainan Pada Sistem Saraf PeriferArtika MayandaPas encore d'évaluation
- 2 - Debilidad AgudaDocument15 pages2 - Debilidad AgudaMarian ZeaPas encore d'évaluation
- Neuromuscular Handout 2006Document12 pagesNeuromuscular Handout 2006Windy MentariiPas encore d'évaluation
- Transverse Myelitis 2013Document60 pagesTransverse Myelitis 2013Habib G. Moutran BarrosoPas encore d'évaluation
- B5W1L9.Peripheral Neuropathy - Lecture Notes 12Document4 pagesB5W1L9.Peripheral Neuropathy - Lecture Notes 12mihalcea alinPas encore d'évaluation
- Neuromusculo Skeletal: Dr. Jumraini Tammasse, SpsDocument46 pagesNeuromusculo Skeletal: Dr. Jumraini Tammasse, SpsFareez HairiPas encore d'évaluation
- Paidepally Srinivas RaoDocument9 pagesPaidepally Srinivas RaoShyam KalavalapalliPas encore d'évaluation
- Muscular DystoniaDocument20 pagesMuscular Dystoniarudresh singhPas encore d'évaluation
- Haramaya University: College of Health and Medical Science Department of Midwifery NeurologyDocument54 pagesHaramaya University: College of Health and Medical Science Department of Midwifery NeurologyMerwan KemalPas encore d'évaluation
- Peripheral NeuropathiesDocument18 pagesPeripheral Neuropathiesjoanna gurtizaPas encore d'évaluation
- Peripheral NeuropathyDocument16 pagesPeripheral NeuropathyAnonymous vnv6QFPas encore d'évaluation
- Neurology, NeuromuscularDisorders-MRyanDocument82 pagesNeurology, NeuromuscularDisorders-MRyanFiras SawanPas encore d'évaluation
- Bio Project T3Document2 pagesBio Project T3Sylvia ChongPas encore d'évaluation
- Cerebellar Disorders: EtiologyDocument3 pagesCerebellar Disorders: EtiologyNistara Singh ChawlaPas encore d'évaluation
- Peripheral Neuropathy Diff Diagnosis and Management AafpDocument6 pagesPeripheral Neuropathy Diff Diagnosis and Management Aafpgus_lions100% (1)
- ParaplegiaDocument5 pagesParaplegiaSampath PavanPas encore d'évaluation
- VEM5384 Clincial Neurology DisordersDocument62 pagesVEM5384 Clincial Neurology DisordersdeadnarwhalPas encore d'évaluation
- Charcot Marrie Tooth DiseaseDocument2 pagesCharcot Marrie Tooth DiseaseAbdur RehmanPas encore d'évaluation
- ATAXIAS PTDocument13 pagesATAXIAS PTKarunya VkPas encore d'évaluation
- Transverse MyelitisDocument19 pagesTransverse MyelitisRainy RaiPas encore d'évaluation
- Myoclonus With DementiaDocument8 pagesMyoclonus With DementiaSantosh DashPas encore d'évaluation
- Nerve Conduction StudiesDocument5 pagesNerve Conduction Studiesأحمد عطيةPas encore d'évaluation
- Peripheral Nerve DisordersDocument33 pagesPeripheral Nerve Disordersbpt2Pas encore d'évaluation
- Amyotrophic Lateral SclerosisDocument11 pagesAmyotrophic Lateral SclerosisAnomalie12345Pas encore d'évaluation
- Thesis About Quack DoctorsDocument22 pagesThesis About Quack DoctorsJervyn Guianan100% (3)
- Dunway Test PaperDocument73 pagesDunway Test PaperShivendu Sharma92% (12)
- Andersen Mcfarlane Wheel-1 2Document17 pagesAndersen Mcfarlane Wheel-1 2api-532703094Pas encore d'évaluation
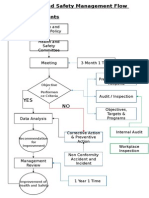
- Health and Safety FlowDocument6 pagesHealth and Safety Flowzaki0304Pas encore d'évaluation
- EY Employee Benefits ManualDocument49 pagesEY Employee Benefits ManualVishnu UdayagiriPas encore d'évaluation
- Kindergarten Physical Education LessonDocument7 pagesKindergarten Physical Education Lessonapi-185987237Pas encore d'évaluation
- Sem 2 M.pharm PresentationDocument16 pagesSem 2 M.pharm PresentationPriya ShahPas encore d'évaluation
- Annual Medical Report Form (DOLE - BWC - HSD - ) H-47-A)Document7 pagesAnnual Medical Report Form (DOLE - BWC - HSD - ) H-47-A)Angelica Ponce100% (2)
- Application of Gis in Health Informatics ResearchDocument6 pagesApplication of Gis in Health Informatics ResearchRaju KoiralaPas encore d'évaluation
- Acute Flaccid Paralysis: Case Investigation FormDocument2 pagesAcute Flaccid Paralysis: Case Investigation FormPaul Angelo E. Caliva0% (1)
- Aute Coronary Syndrome Case ScenarioDocument3 pagesAute Coronary Syndrome Case ScenariokrishcelPas encore d'évaluation
- Hospital ManagementDocument62 pagesHospital ManagementSubramanya DgPas encore d'évaluation
- OHS-PR-09-03-F02 RISK ASSESSMENT For INSTALLATION OF WALL MOUNTED JIB CRANEDocument21 pagesOHS-PR-09-03-F02 RISK ASSESSMENT For INSTALLATION OF WALL MOUNTED JIB CRANEmohammed tofiqPas encore d'évaluation
- QMDocument12 pagesQMGs AbhilashPas encore d'évaluation
- 2018 Attention Deficit Hyperactivity Disorder (ADHD) Controversy Developmental Mechanisms and Multiple Levels of AnalysisDocument28 pages2018 Attention Deficit Hyperactivity Disorder (ADHD) Controversy Developmental Mechanisms and Multiple Levels of AnalysisMarlene LírioPas encore d'évaluation
- An Overview of The Role of Nurses and Midwives in Leadership and Management in EuropeDocument44 pagesAn Overview of The Role of Nurses and Midwives in Leadership and Management in EuropeTCPas encore d'évaluation
- Clinical Practice Guidelines: BY: Ma. Sylvia Emilie B. BeñalesDocument17 pagesClinical Practice Guidelines: BY: Ma. Sylvia Emilie B. Beñalespaulyn ramosPas encore d'évaluation
- Allysa Marie Silbol Beed I1Document3 pagesAllysa Marie Silbol Beed I1Allysa Marie SilbolPas encore d'évaluation
- New Grad ResumeDocument1 pageNew Grad Resumeapi-384352990100% (2)
- Food AdjunctDocument15 pagesFood AdjunctRoby martinus bayaPas encore d'évaluation
- 50 Item Integumentary Exam-BudekDocument8 pages50 Item Integumentary Exam-BudekLj FerolinoPas encore d'évaluation
- Preparation For Child BirthDocument41 pagesPreparation For Child BirthAngelica ErguizaPas encore d'évaluation
- Job Hazard Analysis (JHA) Form - Cyber Sigma1Document1 pageJob Hazard Analysis (JHA) Form - Cyber Sigma1Alvin UbangPas encore d'évaluation
- The Dangers of Sitting - Why Sitting Is The New Smoking - Better Health ChannelDocument10 pagesThe Dangers of Sitting - Why Sitting Is The New Smoking - Better Health ChannelTHE PRATHAM SRIVASTAVA.Pas encore d'évaluation
- The Right To Die: Pre-ReadingDocument8 pagesThe Right To Die: Pre-ReadingGiovanni EscobarPas encore d'évaluation
- Mental Health LetterDocument2 pagesMental Health LetterBethanyPas encore d'évaluation
- Advanced Herd Health Management, Sanitation and HygieneDocument28 pagesAdvanced Herd Health Management, Sanitation and Hygienejane entunaPas encore d'évaluation
- CORAD 101, Health and Welfare ProgramsDocument5 pagesCORAD 101, Health and Welfare ProgramsdaybudsvaldezPas encore d'évaluation
- Nutrition For Liver Gallbladder and Pancreas DiseasesDocument39 pagesNutrition For Liver Gallbladder and Pancreas DiseasesMaricar Car CartallaPas encore d'évaluation
- CDEM 17jan Complete WEBDocument24 pagesCDEM 17jan Complete WEBRicardo Jonathan Ayala GarciaPas encore d'évaluation
- Love Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)D'EverandLove Life: How to Raise Your Standards, Find Your Person, and Live Happily (No Matter What)Évaluation : 3 sur 5 étoiles3/5 (1)
- ADHD is Awesome: A Guide to (Mostly) Thriving with ADHDD'EverandADHD is Awesome: A Guide to (Mostly) Thriving with ADHDÉvaluation : 5 sur 5 étoiles5/5 (2)
- The Age of Magical Overthinking: Notes on Modern IrrationalityD'EverandThe Age of Magical Overthinking: Notes on Modern IrrationalityÉvaluation : 4 sur 5 étoiles4/5 (29)
- LIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionD'EverandLIT: Life Ignition Tools: Use Nature's Playbook to Energize Your Brain, Spark Ideas, and Ignite ActionÉvaluation : 4 sur 5 étoiles4/5 (404)
- By the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsD'EverandBy the Time You Read This: The Space between Cheslie's Smile and Mental Illness—Her Story in Her Own WordsPas encore d'évaluation
- Summary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedD'EverandSummary: The Psychology of Money: Timeless Lessons on Wealth, Greed, and Happiness by Morgan Housel: Key Takeaways, Summary & Analysis IncludedÉvaluation : 5 sur 5 étoiles5/5 (81)
- Think This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeD'EverandThink This, Not That: 12 Mindshifts to Breakthrough Limiting Beliefs and Become Who You Were Born to BeÉvaluation : 2 sur 5 étoiles2/5 (1)
- Mindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessD'EverandMindset by Carol S. Dweck - Book Summary: The New Psychology of SuccessÉvaluation : 4.5 sur 5 étoiles4.5/5 (328)
- The Obesity Code: Unlocking the Secrets of Weight LossD'EverandThe Obesity Code: Unlocking the Secrets of Weight LossÉvaluation : 4 sur 5 étoiles4/5 (6)
- The Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaD'EverandThe Body Keeps the Score by Bessel Van der Kolk, M.D. - Book Summary: Brain, Mind, and Body in the Healing of TraumaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- Summary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisD'EverandSummary: Outlive: The Science and Art of Longevity by Peter Attia MD, With Bill Gifford: Key Takeaways, Summary & AnalysisÉvaluation : 4.5 sur 5 étoiles4.5/5 (42)
- The Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsD'EverandThe Ritual Effect: From Habit to Ritual, Harness the Surprising Power of Everyday ActionsÉvaluation : 3.5 sur 5 étoiles3.5/5 (3)
- Raising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsD'EverandRaising Mentally Strong Kids: How to Combine the Power of Neuroscience with Love and Logic to Grow Confident, Kind, Responsible, and Resilient Children and Young AdultsÉvaluation : 5 sur 5 étoiles5/5 (1)
- The Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeD'EverandThe Courage Habit: How to Accept Your Fears, Release the Past, and Live Your Courageous LifeÉvaluation : 4.5 sur 5 étoiles4.5/5 (253)
- The Comfort of Crows: A Backyard YearD'EverandThe Comfort of Crows: A Backyard YearÉvaluation : 4.5 sur 5 étoiles4.5/5 (23)
- Dark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.D'EverandDark Psychology & Manipulation: Discover How To Analyze People and Master Human Behaviour Using Emotional Influence Techniques, Body Language Secrets, Covert NLP, Speed Reading, and Hypnosis.Évaluation : 4.5 sur 5 étoiles4.5/5 (110)
- Why We Die: The New Science of Aging and the Quest for ImmortalityD'EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityÉvaluation : 4 sur 5 étoiles4/5 (5)
- Outlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisD'EverandOutlive: The Science and Art of Longevity by Peter Attia: Key Takeaways, Summary & AnalysisÉvaluation : 4 sur 5 étoiles4/5 (1)
- Cult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryD'EverandCult, A Love Story: Ten Years Inside a Canadian Cult and the Subsequent Long Road of RecoveryÉvaluation : 4 sur 5 étoiles4/5 (45)
- Raising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsD'EverandRaising Good Humans: A Mindful Guide to Breaking the Cycle of Reactive Parenting and Raising Kind, Confident KidsÉvaluation : 4.5 sur 5 étoiles4.5/5 (170)
- Summary: Thinking, Fast and Slow: by Daniel Kahneman: Key Takeaways, Summary & Analysis IncludedD'EverandSummary: Thinking, Fast and Slow: by Daniel Kahneman: Key Takeaways, Summary & Analysis IncludedÉvaluation : 4 sur 5 étoiles4/5 (61)
- Manipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesD'EverandManipulation: The Ultimate Guide To Influence People with Persuasion, Mind Control and NLP With Highly Effective Manipulation TechniquesÉvaluation : 4.5 sur 5 étoiles4.5/5 (1412)
- How to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingD'EverandHow to ADHD: The Ultimate Guide and Strategies for Productivity and Well-BeingÉvaluation : 1 sur 5 étoiles1/5 (1)
- The Marshmallow Test: Mastering Self-ControlD'EverandThe Marshmallow Test: Mastering Self-ControlÉvaluation : 4.5 sur 5 étoiles4.5/5 (59)