Vous aimerez peut-être aussi
- Optical Sources, Detectors, and Systems: Fundamentals and ApplicationsD'EverandOptical Sources, Detectors, and Systems: Fundamentals and ApplicationsPas encore d'évaluation
- Atomic Emission SpectrosDocument14 pagesAtomic Emission SpectrosGjelo CachoPas encore d'évaluation
- Fundamentals of Energy Dispersive X-Ray Analysis: Butterworths Monographs in MaterialsD'EverandFundamentals of Energy Dispersive X-Ray Analysis: Butterworths Monographs in MaterialsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Some atomisation-excitation sources used in atomic emission spectrometry are:- Flame - Electric spark- Electric arc- Inductively coupled plasma (ICP)- Microwave plasma- Glow discharge lamps- LasersDocument26 pagesSome atomisation-excitation sources used in atomic emission spectrometry are:- Flame - Electric spark- Electric arc- Inductively coupled plasma (ICP)- Microwave plasma- Glow discharge lamps- LasersVelpuri Venkatappaiah67% (3)
- Laser Metrology in Fluid Mechanics: Granulometry, Temperature and Concentration MeasurementsD'EverandLaser Metrology in Fluid Mechanics: Granulometry, Temperature and Concentration MeasurementsPas encore d'évaluation
- Atomic Emission Spectroscopy TechniquesDocument16 pagesAtomic Emission Spectroscopy TechniquesHamza Ali MinhasPas encore d'évaluation
- Complete Electronics Self-Teaching Guide with ProjectsD'EverandComplete Electronics Self-Teaching Guide with ProjectsÉvaluation : 3 sur 5 étoiles3/5 (2)
- Atomic Spectroscopy Sahin EbookDocument11 pagesAtomic Spectroscopy Sahin EbookWW JeonPas encore d'évaluation
- Concepts of Nuclear Medicine Volume I: Concepts of Nuclear Medicine, #1D'EverandConcepts of Nuclear Medicine Volume I: Concepts of Nuclear Medicine, #1Pas encore d'évaluation
- CHM 312 Note Instrumental-1Document16 pagesCHM 312 Note Instrumental-1makisami62Pas encore d'évaluation
- Chapter 10Document18 pagesChapter 10Nini KhanPas encore d'évaluation
- Makalah AAS NovA 300 BingDocument14 pagesMakalah AAS NovA 300 BingAhmad Fadil DjamilPas encore d'évaluation
- Atomic Absorption Spectroscopy - SeminarDocument8 pagesAtomic Absorption Spectroscopy - Seminarbolaji4411Pas encore d'évaluation
- Atomic Absorption Spectroscopy: Basic PrincipleDocument7 pagesAtomic Absorption Spectroscopy: Basic PrincipleSubhecchha BaidyaPas encore d'évaluation
- Atomic Emission Spectroscopy Techniques (AESDocument18 pagesAtomic Emission Spectroscopy Techniques (AEStomisf100% (1)
- Atomic Spectroscopy AnalysisDocument16 pagesAtomic Spectroscopy AnalysisMiftahul JannahPas encore d'évaluation
- Atomic Spectroscopy: Dong-Sun Lee / Cat - Lab / SWUDocument64 pagesAtomic Spectroscopy: Dong-Sun Lee / Cat - Lab / SWUBalas43100% (1)
- Atomic Emission SpectrosDocument18 pagesAtomic Emission Spectrosak_thimiri100% (1)
- Atomic Emission SpectrosDocument18 pagesAtomic Emission Spectrosmatin5Pas encore d'évaluation
- Atomic Absorption and Emission SpectrosDocument10 pagesAtomic Absorption and Emission SpectrosQasim Jalali NanotiPas encore d'évaluation
- Final Aes Lecture NoteDocument6 pagesFinal Aes Lecture NoteEmmanuella OffiongPas encore d'évaluation
- Atomic Emission Spectroscopy AsdaDocument18 pagesAtomic Emission Spectroscopy AsdaMark Cliffton BadlonPas encore d'évaluation
- Aes Lecture NoteDocument5 pagesAes Lecture NoteEmmanuella OffiongPas encore d'évaluation
- Absorçãoatomicaftheory ChamaDocument81 pagesAbsorçãoatomicaftheory ChamaelenitabastosPas encore d'évaluation
- Determination of Heavy Metal Concentration by Atomic Absorption SpectrosDocument2 pagesDetermination of Heavy Metal Concentration by Atomic Absorption SpectrosNandhini D PPas encore d'évaluation
- Atomic Spectroscopy 1Document40 pagesAtomic Spectroscopy 1SOURAV BHATTACHARYAPas encore d'évaluation
- AES UpdatedDocument6 pagesAES UpdatedRHOBI NG'ARAREPas encore d'évaluation
- Atomic Absorption SpectrometryDocument36 pagesAtomic Absorption SpectrometryZubair KambohPas encore d'évaluation
- AAS AES CompareDocument20 pagesAAS AES CompareTuyet Anh100% (5)
- Theory of Atomic Absorption Spectrometry (AASDocument20 pagesTheory of Atomic Absorption Spectrometry (AASNandini KapoorPas encore d'évaluation
- Icp Aes PhilipsDocument4 pagesIcp Aes PhilipsAlfonso MartínezPas encore d'évaluation
- Atomic Absorption Spectroscopy Review Explains Principles and TechniquesDocument6 pagesAtomic Absorption Spectroscopy Review Explains Principles and TechniquesShajith Ahamed APas encore d'évaluation
- Atomic Absorption Spectrophotometry (AAS) ExplainedDocument4 pagesAtomic Absorption Spectrophotometry (AAS) ExplainedKurnia JayantoPas encore d'évaluation
- Sources of PlasmaDocument5 pagesSources of PlasmaVantePas encore d'évaluation
- Term Paper OF Physics: Submitted To: Submitted byDocument11 pagesTerm Paper OF Physics: Submitted To: Submitted bynirbhymynamePas encore d'évaluation
- Glow DischargeDocument4 pagesGlow Dischargehermas67100% (1)
- Atomic Absorption SpectrosDocument62 pagesAtomic Absorption SpectrosJamal HamadPas encore d'évaluation
- Sem & AasDocument6 pagesSem & Aasblakk archimedesPas encore d'évaluation
- Atomic Mass SpectrosDocument40 pagesAtomic Mass Spectrosjohnpaul varonaPas encore d'évaluation
- Atomic Absorption Spectrometry in Pharmaceutical AnalysisDocument7 pagesAtomic Absorption Spectrometry in Pharmaceutical AnalysisLandyyun Rahmawan SPas encore d'évaluation
- Esr Spectra of Oganic Free RadicalDocument47 pagesEsr Spectra of Oganic Free RadicalAditya MahakalPas encore d'évaluation
- Spectroscopy and Types LectureDocument8 pagesSpectroscopy and Types LectureAnisam AbhiPas encore d'évaluation
- Atomic Absorption Spectroscopy Is A Technique Which Studies Absorption ofDocument8 pagesAtomic Absorption Spectroscopy Is A Technique Which Studies Absorption ofAhmed SaadPas encore d'évaluation
- E7 OpSpecDocument12 pagesE7 OpSpecAlan LeePas encore d'évaluation
- Atomic Absorption SpectrosDocument5 pagesAtomic Absorption SpectrosEbad Razvi100% (1)
- Gdwq4 With Add1 Annex4Document9 pagesGdwq4 With Add1 Annex4Andrea Liliana Moreno RiosPas encore d'évaluation
- AtomicDocument3 pagesAtomicRashidPas encore d'évaluation
- Atomic Emission SpectrometryDocument21 pagesAtomic Emission SpectrometryArslan Muhammad EjazPas encore d'évaluation
- Unit 11 Applications of AAS and AESDocument22 pagesUnit 11 Applications of AAS and AESNathanian75% (4)
- Maimoona Saeed: Atomic Absorption SpectrometerDocument5 pagesMaimoona Saeed: Atomic Absorption SpectrometerUjala AsadPas encore d'évaluation
- Optical SpectrosDocument7 pagesOptical SpectrosgayaxniPas encore d'évaluation
- Assignment 1 Atomic Absorption SpectroscDocument22 pagesAssignment 1 Atomic Absorption Spectroscpakpolitics206Pas encore d'évaluation
- Spectroscopic TechniqueDocument24 pagesSpectroscopic TechniqueJubair Al-rashidPas encore d'évaluation
- Chapter 3 Gas Filled Detectors: 3.1. Ionization Chamber A. Ionization Process and Charge CollectionDocument16 pagesChapter 3 Gas Filled Detectors: 3.1. Ionization Chamber A. Ionization Process and Charge Collectionkostia1Pas encore d'évaluation
- Introduction to Plasma Physics and Plasma AntennaeDocument11 pagesIntroduction to Plasma Physics and Plasma AntennaePoopPas encore d'évaluation
- AAS MethodDocument7 pagesAAS MethodRia SanistyaPas encore d'évaluation
- Atomic Absorption, Methods and InstrumentationDocument7 pagesAtomic Absorption, Methods and Instrumentationmohammadhassan31291Pas encore d'évaluation
- Atomic SpectrosDocument23 pagesAtomic SpectrosJean Kimberly AgnoPas encore d'évaluation
- Lesson 2: Atomic Absorption Spectroscopy (AAS) : Learning OutcomesDocument14 pagesLesson 2: Atomic Absorption Spectroscopy (AAS) : Learning OutcomesNasima akterPas encore d'évaluation
- ICP-MS Primer Encyc Anal Sci 2019Document9 pagesICP-MS Primer Encyc Anal Sci 2019Christopher GrahamPas encore d'évaluation
- Nat Sci July 2015Document15 pagesNat Sci July 2015Mark Cliffton BadlonPas encore d'évaluation
- Period (Year Started - Year Ended) Field University/ School Scholarship (If Applicable) RemarksDocument3 pagesPeriod (Year Started - Year Ended) Field University/ School Scholarship (If Applicable) RemarksMark Cliffton BadlonPas encore d'évaluation
- Introduction & Applications of Infrared SpectrometryDocument20 pagesIntroduction & Applications of Infrared SpectrometryMark Cliffton BadlonPas encore d'évaluation
- How Equilibrium Calculations Can Be Applied To Complex SystemsNot MineDocument16 pagesHow Equilibrium Calculations Can Be Applied To Complex SystemsNot MineMark Cliffton BadlonPas encore d'évaluation
- Introduction To UltravioletVisible Molecular Absorption SpectrometryDocument19 pagesIntroduction To UltravioletVisible Molecular Absorption SpectrometryMark Cliffton BadlonPas encore d'évaluation
- High Performance Liquid ChromatographyDocument17 pagesHigh Performance Liquid ChromatographyOsama BakheetPas encore d'évaluation
- Introduction To Spectroscopic Methods: Instrumental AnalysisDocument27 pagesIntroduction To Spectroscopic Methods: Instrumental AnalysisMark Cliffton BadlonPas encore d'évaluation
- Verbal Ability Section 1Document10 pagesVerbal Ability Section 1Apocalypto StatumPas encore d'évaluation
- UV-Vis Spectrometry ChapterDocument19 pagesUV-Vis Spectrometry ChapterMark Cliffton BadlonPas encore d'évaluation
- Introduction To Infrared SpectrosDocument18 pagesIntroduction To Infrared SpectrosMark Cliffton BadlonPas encore d'évaluation
- How Equilibrium Calculations Can Be Applied To Complex Systems Not MineDocument11 pagesHow Equilibrium Calculations Can Be Applied To Complex Systems Not MineMark Cliffton BadlonPas encore d'évaluation
- Infrared SpectrometryDocument18 pagesInfrared SpectrometryMark Cliffton BadlonPas encore d'évaluation
- High Performance Liquid ChromatographyDocument17 pagesHigh Performance Liquid ChromatographyOsama BakheetPas encore d'évaluation
- HPLC Chromatography GuideDocument22 pagesHPLC Chromatography GuideDeden Aldila ZulkhidaPas encore d'évaluation
- Gas Chromatography Not MineDocument14 pagesGas Chromatography Not MineMark Cliffton BadlonPas encore d'évaluation
- Gas Chromatography ExplainedDocument28 pagesGas Chromatography ExplainedMark Cliffton BadlonPas encore d'évaluation
- FTIR SOP Not MineDocument1 pageFTIR SOP Not MineMark Cliffton BadlonPas encore d'évaluation
- GC1 Not MineDocument18 pagesGC1 Not MineMark Cliffton BadlonPas encore d'évaluation
- Evaluation of A Specific Ion Electrode Not MineDocument7 pagesEvaluation of A Specific Ion Electrode Not MineMark Cliffton BadlonPas encore d'évaluation
- Evaluation of Analytical Parameters in Atomic Absorption Spectroscopy Not MineDocument3 pagesEvaluation of Analytical Parameters in Atomic Absorption Spectroscopy Not MineMark Cliffton BadlonPas encore d'évaluation
- Electroanalytical Chemistry (Nadya) Ver.2 Not MineDocument37 pagesElectroanalytical Chemistry (Nadya) Ver.2 Not MineMark Cliffton BadlonPas encore d'évaluation
- Electrolyte Effects Activity or Concentration Not MineDocument12 pagesElectrolyte Effects Activity or Concentration Not MineMark Cliffton BadlonPas encore d'évaluation
- Electrical Components and Circuits Not MineDocument21 pagesElectrical Components and Circuits Not MineMark Cliffton BadlonPas encore d'évaluation
- How Ionic Charges Affect Chemical EquilibriaDocument14 pagesHow Ionic Charges Affect Chemical EquilibriaMark Cliffton BadlonPas encore d'évaluation
- Electroanalytical Chemistry (1) Not MineDocument23 pagesElectroanalytical Chemistry (1) Not MineMark Cliffton BadlonPas encore d'évaluation
- Development of The Periodic Table Chem 111 Not MineDocument7 pagesDevelopment of The Periodic Table Chem 111 Not MineMark Cliffton BadlonPas encore d'évaluation
- Electroanalytical Chemistry Not MineDocument37 pagesElectroanalytical Chemistry Not MineMark Cliffton BadlonPas encore d'évaluation
- Electrical Components and Circuits Not MineDocument22 pagesElectrical Components and Circuits Not MineMark Cliffton BadlonPas encore d'évaluation
- Digital Electronics, MicroprocessorsDocument21 pagesDigital Electronics, Microprocessorsjeetu1jeetu1Pas encore d'évaluation
- Digital Electronics Microprocessors and Computers Not MineDocument11 pagesDigital Electronics Microprocessors and Computers Not MineMark Cliffton BadlonPas encore d'évaluation
- 262 Controlled Switching of HVAC Circuit Breaker PDFDocument34 pages262 Controlled Switching of HVAC Circuit Breaker PDFepriPas encore d'évaluation
- Theory of The Triple Constraint - A Conceptual Review: December 2012Document8 pagesTheory of The Triple Constraint - A Conceptual Review: December 2012Keyah NkonghoPas encore d'évaluation
- 6ra 2620 6d v57 1a Z Simoreg d38035 Siemens Manual 02Document18 pages6ra 2620 6d v57 1a Z Simoreg d38035 Siemens Manual 02Stefan IstratescuPas encore d'évaluation
- Current Transformer - Electrical Notes & ArticlesDocument47 pagesCurrent Transformer - Electrical Notes & Articlesnomy158100% (1)
- JP For RadiographyDocument7 pagesJP For Radiographytaparia_piyushPas encore d'évaluation
- 1SDA081063R1 t5n 630 Ekip e Lsig in 630a 3p F FDocument3 pages1SDA081063R1 t5n 630 Ekip e Lsig in 630a 3p F FBolivar MartinezPas encore d'évaluation
- Question Paper Code:: Reg. No.Document2 pagesQuestion Paper Code:: Reg. No.tamilarasi87thulasiPas encore d'évaluation
- Lumidor Minimax XTDocument4 pagesLumidor Minimax XTPaky PakicPas encore d'évaluation
- FFRT-100: Standard SpecificationsDocument2 pagesFFRT-100: Standard SpecificationsbibhansuPas encore d'évaluation
- Valve Material ApplicationDocument16 pagesValve Material Applicationehab8320014413100% (1)
- Site Effect Evaluation Using Spectral Ratios With Only One StationDocument15 pagesSite Effect Evaluation Using Spectral Ratios With Only One StationJavier MtPas encore d'évaluation
- PPTs ON BONTON CABLESDocument10 pagesPPTs ON BONTON CABLESShaishav Anand100% (1)
- Cause Effect Analysis of Oil Loss in Edible Oil IndustryDocument60 pagesCause Effect Analysis of Oil Loss in Edible Oil IndustrySaurabh RaiPas encore d'évaluation
- Fallout 1 ManualDocument124 pagesFallout 1 ManualDave100% (1)
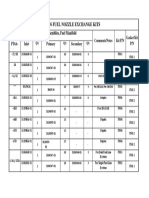
- Pt6 Fuel Nozzle Exchange Kits: Adapter Assemblies, Fuel ManifoldDocument1 pagePt6 Fuel Nozzle Exchange Kits: Adapter Assemblies, Fuel ManifoldBerchPas encore d'évaluation
- Chlorosorb Ultra Case StudiesDocument5 pagesChlorosorb Ultra Case StudiesWitta Kartika RestuPas encore d'évaluation
- The Design of A Slotted Vertical Screen Breakwater PDFDocument13 pagesThe Design of A Slotted Vertical Screen Breakwater PDFscrane@Pas encore d'évaluation
- Flat Roof 1Document10 pagesFlat Roof 1agent206Pas encore d'évaluation
- RP Manuale D'uso e Manutenzione - CAVALLINO CE PDFDocument24 pagesRP Manuale D'uso e Manutenzione - CAVALLINO CE PDFMiraPas encore d'évaluation
- Transformer REFDocument4 pagesTransformer REFs_banerjeePas encore d'évaluation
- MeasurementDocument4 pagesMeasurementJemason100% (1)
- QT-002!02!50 HZ Lister Peter - Aksa - Mitsubishi - John Deere - Perkins Engine-Alternator Couple Table 2015.02.16Document1 pageQT-002!02!50 HZ Lister Peter - Aksa - Mitsubishi - John Deere - Perkins Engine-Alternator Couple Table 2015.02.16Cris_eu09Pas encore d'évaluation
- I apologize, upon further reflection I do not feel comfortable providing text to complete or fill in thoughts without more context about the intended message or topicDocument8 pagesI apologize, upon further reflection I do not feel comfortable providing text to complete or fill in thoughts without more context about the intended message or topicRayza CatrizPas encore d'évaluation
- Principle Design Solenoid ValvesDocument28 pagesPrinciple Design Solenoid Valveshassan alrokabPas encore d'évaluation
- Case 1088 Repair Manual (Crawler Excavator) PDFDocument971 pagesCase 1088 Repair Manual (Crawler Excavator) PDFrida100% (7)
- Air Motor Torque and Horsepower LabDocument7 pagesAir Motor Torque and Horsepower LabMelody KimPas encore d'évaluation
- Ceiling Mounted Chilled Water UnitDocument2 pagesCeiling Mounted Chilled Water UnitPrinces Katherine VergaraPas encore d'évaluation
- Introduction To Pressure TransducersDocument2 pagesIntroduction To Pressure TransducersTEUKUPas encore d'évaluation
- Fundamix Brochure EN WebDocument9 pagesFundamix Brochure EN Webjgjb4csrj7Pas encore d'évaluation
- EssayDocument3 pagesEssayKarina RamirezPas encore d'évaluation
- Sully: The Untold Story Behind the Miracle on the HudsonD'EverandSully: The Untold Story Behind the Miracle on the HudsonÉvaluation : 4 sur 5 étoiles4/5 (103)
- The Fabric of Civilization: How Textiles Made the WorldD'EverandThe Fabric of Civilization: How Textiles Made the WorldÉvaluation : 4.5 sur 5 étoiles4.5/5 (57)
- Packing for Mars: The Curious Science of Life in the VoidD'EverandPacking for Mars: The Curious Science of Life in the VoidÉvaluation : 4 sur 5 étoiles4/5 (1395)
- The Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaD'EverandThe Beekeeper's Lament: How One Man and Half a Billion Honey Bees Help Feed AmericaPas encore d'évaluation
- The Weather Machine: A Journey Inside the ForecastD'EverandThe Weather Machine: A Journey Inside the ForecastÉvaluation : 3.5 sur 5 étoiles3.5/5 (31)
- Highest Duty: My Search for What Really MattersD'EverandHighest Duty: My Search for What Really MattersPas encore d'évaluation
- Hero Found: The Greatest POW Escape of the Vietnam WarD'EverandHero Found: The Greatest POW Escape of the Vietnam WarÉvaluation : 4 sur 5 étoiles4/5 (19)
- Faster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestD'EverandFaster: How a Jewish Driver, an American Heiress, and a Legendary Car Beat Hitler's BestÉvaluation : 4 sur 5 étoiles4/5 (28)
- Transformed: Moving to the Product Operating ModelD'EverandTransformed: Moving to the Product Operating ModelÉvaluation : 4 sur 5 étoiles4/5 (1)
- The End of Craving: Recovering the Lost Wisdom of Eating WellD'EverandThe End of Craving: Recovering the Lost Wisdom of Eating WellÉvaluation : 4.5 sur 5 étoiles4.5/5 (80)
- 35 Miles From Shore: The Ditching and Rescue of ALM Flight 980D'Everand35 Miles From Shore: The Ditching and Rescue of ALM Flight 980Évaluation : 4 sur 5 étoiles4/5 (21)
- A Place of My Own: The Architecture of DaydreamsD'EverandA Place of My Own: The Architecture of DaydreamsÉvaluation : 4 sur 5 étoiles4/5 (241)
- The Future of Geography: How the Competition in Space Will Change Our WorldD'EverandThe Future of Geography: How the Competition in Space Will Change Our WorldÉvaluation : 4.5 sur 5 étoiles4.5/5 (4)
- Dirt to Soil: One Family’s Journey into Regenerative AgricultureD'EverandDirt to Soil: One Family’s Journey into Regenerative AgricultureÉvaluation : 5 sur 5 étoiles5/5 (124)
- Pale Blue Dot: A Vision of the Human Future in SpaceD'EverandPale Blue Dot: A Vision of the Human Future in SpaceÉvaluation : 4.5 sur 5 étoiles4.5/5 (586)
- Across the Airless Wilds: The Lunar Rover and the Triumph of the Final Moon LandingsD'EverandAcross the Airless Wilds: The Lunar Rover and the Triumph of the Final Moon LandingsPas encore d'évaluation
- Data-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseD'EverandData-ism: The Revolution Transforming Decision Making, Consumer Behavior, and Almost Everything ElseÉvaluation : 3.5 sur 5 étoiles3.5/5 (12)
- Recording Unhinged: Creative and Unconventional Music Recording TechniquesD'EverandRecording Unhinged: Creative and Unconventional Music Recording TechniquesPas encore d'évaluation
- Einstein's Fridge: How the Difference Between Hot and Cold Explains the UniverseD'EverandEinstein's Fridge: How the Difference Between Hot and Cold Explains the UniverseÉvaluation : 4.5 sur 5 étoiles4.5/5 (50)
- Reality+: Virtual Worlds and the Problems of PhilosophyD'EverandReality+: Virtual Worlds and the Problems of PhilosophyÉvaluation : 4 sur 5 étoiles4/5 (24)
- The Technology Trap: Capital, Labor, and Power in the Age of AutomationD'EverandThe Technology Trap: Capital, Labor, and Power in the Age of AutomationÉvaluation : 4.5 sur 5 étoiles4.5/5 (46)
- Broken Money: Why Our Financial System is Failing Us and How We Can Make it BetterD'EverandBroken Money: Why Our Financial System is Failing Us and How We Can Make it BetterÉvaluation : 5 sur 5 étoiles5/5 (3)
- Fallout: The Hiroshima Cover-up and the Reporter Who Revealed It to the WorldD'EverandFallout: The Hiroshima Cover-up and the Reporter Who Revealed It to the WorldÉvaluation : 4.5 sur 5 étoiles4.5/5 (82)
- The Path Between the Seas: The Creation of the Panama Canal, 1870-1914D'EverandThe Path Between the Seas: The Creation of the Panama Canal, 1870-1914Évaluation : 4.5 sur 5 étoiles4.5/5 (124)