Vous aimerez peut-être aussi
- Mutación Cromosómica, Variaciones e HibridosDocument16 pagesMutación Cromosómica, Variaciones e HibridosluciaPas encore d'évaluation
- CROMOSOPATIADocument23 pagesCROMOSOPATIASamir MirandaPas encore d'évaluation
- Diferencias Hombre Mujer: Descubre Los Últimos Hallazgos Científicos Sobre Las Diferencias Entre Mujeres Y HombresD'EverandDiferencias Hombre Mujer: Descubre Los Últimos Hallazgos Científicos Sobre Las Diferencias Entre Mujeres Y HombresPas encore d'évaluation
- Anomalías CromosomáticasDocument6 pagesAnomalías CromosomáticasErnesto Erazo PortalPas encore d'évaluation
- Citogenitca HumanaDocument24 pagesCitogenitca HumanaJose Andres Castro NavarroPas encore d'évaluation
- Gentica HumanaDocument14 pagesGentica HumanaJair Gomez GuevaraPas encore d'évaluation
- Cariotipo HumanoDocument14 pagesCariotipo HumanoRafael LeonPas encore d'évaluation
- Comentarios - "Alteraciones Cromosómicas"Document9 pagesComentarios - "Alteraciones Cromosómicas"AGUILAR HERNANDEZ MARIANA ITZELPas encore d'évaluation
- CromosomopatíasDocument14 pagesCromosomopatíasFranco ContreraPas encore d'évaluation
- Mutación CromosómicaDocument4 pagesMutación CromosómicaFLOR DE MARIA ROSARIO HUARCAYA LIMACHIPas encore d'évaluation
- Informe IngrithDocument15 pagesInforme IngrithElGatoDeAuronplayPas encore d'évaluation
- Alteraciones CromosómicasDocument3 pagesAlteraciones CromosómicasRoland M ReyesPas encore d'évaluation
- Anomalías CromosomicasDocument5 pagesAnomalías Cromosomicaslucia floresPas encore d'évaluation
- Herencia CromosómicaDocument6 pagesHerencia CromosómicaNicolas RodriguezPas encore d'évaluation
- Anomalias CromosomicasDocument15 pagesAnomalias Cromosomicasanthony lopezPas encore d'évaluation
- Sesion 2 Anomalias Cromosomicas PDFDocument12 pagesSesion 2 Anomalias Cromosomicas PDFMilagros Blas PizarroPas encore d'évaluation
- Mutaciones Cromosomicas!Document30 pagesMutaciones Cromosomicas!Anonymous ApyNcvV2Vl100% (1)
- CosmicasDocument8 pagesCosmicasMaríaFernanda VasquezPas encore d'évaluation
- Cromosomas Normales, Alteraciones en Numero Estructura y SindromesDocument6 pagesCromosomas Normales, Alteraciones en Numero Estructura y SindromesLuis Antonio MartinézPas encore d'évaluation
- Las Enfermedades Cromosómicas Están Provocadas Por Anomalías CromosómicasDocument14 pagesLas Enfermedades Cromosómicas Están Provocadas Por Anomalías CromosómicasCYPHER 4 :vPas encore d'évaluation
- TRIPLOIDÍADocument6 pagesTRIPLOIDÍAvelichek67% (3)
- Presentación Medicina Embriología Profesional Beige GranateDocument17 pagesPresentación Medicina Embriología Profesional Beige GranateMERCEDES ANYPSA PALACIOS ROMANPas encore d'évaluation
- MutacionesDocument47 pagesMutacionesCristian Soto PeñaililloPas encore d'évaluation
- Anomalías CromosómicasDocument6 pagesAnomalías CromosómicasReyna LopezPas encore d'évaluation
- Citogenética Básica y Anomalias CromosómicasDocument108 pagesCitogenética Básica y Anomalias CromosómicasAlejandra Gonzalez100% (1)
- Tema16 231218174850 5b47f7d1Document82 pagesTema16 231218174850 5b47f7d1Samy SandovalPas encore d'évaluation
- Anomalías CromosómicasDocument14 pagesAnomalías Cromosómicaspipidel82Pas encore d'évaluation
- Cariotipo AnormalDocument39 pagesCariotipo Anormaljohnnyr27Pas encore d'évaluation
- Alteraciones Cromosomicas IIDocument7 pagesAlteraciones Cromosomicas IIKarla Veneros TerronesPas encore d'évaluation
- Clase de Citogenética HumanaDocument62 pagesClase de Citogenética HumanaPaola FloresPas encore d'évaluation
- Enfermedades CromosomicasDocument14 pagesEnfermedades Cromosomicassebastian andres sepulveda de leonPas encore d'évaluation
- Anomalías CromosómicasDocument88 pagesAnomalías CromosómicasTalulah GoshPas encore d'évaluation
- Alteraciones CromosomicasDocument75 pagesAlteraciones CromosomicasAxel QuispePas encore d'évaluation
- AberracionesDocument47 pagesAberracionesCatherine Cinthya RIMAC PANEZPas encore d'évaluation
- Anormalidades Cromosómicas en HumanosDocument38 pagesAnormalidades Cromosómicas en HumanosGoku SaPas encore d'évaluation
- Aberraciones Cromosomicas.Document12 pagesAberraciones Cromosomicas.Brandon CLPas encore d'évaluation
- Alteraciones Cromosomicas2Document16 pagesAlteraciones Cromosomicas2Hiciab Luismi CabreraPas encore d'évaluation
- GENÉTICA Segundo ParcialDocument11 pagesGENÉTICA Segundo ParcialAfrica ReynosoPas encore d'évaluation
- TP 4 CariotipoDocument8 pagesTP 4 CariotipoKevin AyllonPas encore d'évaluation
- Clase 5 MutacionesDocument42 pagesClase 5 MutacionesJuan Carlos Chamorro ElizondoPas encore d'évaluation
- Mosaicismo CromosomicoDocument6 pagesMosaicismo CromosomicoJhonatan Fernando Salcedo OrtegaPas encore d'évaluation
- Enfermedades Producidas Por La Alteración de CromosomasDocument8 pagesEnfermedades Producidas Por La Alteración de CromosomasCynthia MongelosPas encore d'évaluation
- Cariotipo HumanoDocument41 pagesCariotipo HumanoAlejandra MelendezPas encore d'évaluation
- Anomalías Anatómicas Congénitas o Defectos Del NacimientoDocument4 pagesAnomalías Anatómicas Congénitas o Defectos Del NacimientoHarizel EscalonaPas encore d'évaluation
- Citogenetica HumanaDocument77 pagesCitogenetica HumanaJesus Francisco Flores Delgado100% (1)
- Seminario 1 Embriología: Alteraciones CromosomicasDocument46 pagesSeminario 1 Embriología: Alteraciones Cromosomicassebastian perez67% (3)
- Seminario 12 Anomalias CromosomicasDocument22 pagesSeminario 12 Anomalias CromosomicasLeslie Zavaleta BazanPas encore d'évaluation
- Alteraciones CromosomicasDocument21 pagesAlteraciones CromosomicasJonathan GutierrezPas encore d'évaluation
- Trabajo de TRANSTORNOS CROMOSOMAS - YAJAIDADocument2 pagesTrabajo de TRANSTORNOS CROMOSOMAS - YAJAIDARosa Maldonado MartinezPas encore d'évaluation
- Defectos Congenitos y Abortos Expontaneos Factores Cromosomicos y GeneticosDocument6 pagesDefectos Congenitos y Abortos Expontaneos Factores Cromosomicos y GeneticosDavidCumbicusPas encore d'évaluation
- Mutaciones Bioquímicas o Nutritivas MARIANGELDocument5 pagesMutaciones Bioquímicas o Nutritivas MARIANGELKarl GreenPas encore d'évaluation
- Anomalías Cromosómicas EstructuralesDocument7 pagesAnomalías Cromosómicas EstructuralesIvan R-RosarioPas encore d'évaluation
- Cariotipo HumanoDocument13 pagesCariotipo HumanoRicardo CrisóstomoPas encore d'évaluation
- TRASTORNOS CROMOSOMICOS UmbDocument23 pagesTRASTORNOS CROMOSOMICOS UmbManu SaurioPas encore d'évaluation
- TRISOMÍAS ResumenDocument3 pagesTRISOMÍAS ResumenAngela DávilaPas encore d'évaluation
- Clasificación de CromosomasDocument9 pagesClasificación de CromosomasCruzMilagrosQuispeLopezPas encore d'évaluation
- Monografía Foro 8Document24 pagesMonografía Foro 8cesar davidPas encore d'évaluation
- Ciencias - 2do 27 04Document8 pagesCiencias - 2do 27 04Silvina EncinaPas encore d'évaluation
- Agentes Práctico Final Trabajo 3Document2 pagesAgentes Práctico Final Trabajo 3Darwin HerreraPas encore d'évaluation
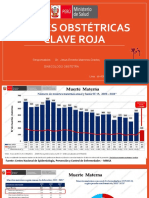
- Claves Obstetricas Clave Roja 2021Document31 pagesClaves Obstetricas Clave Roja 2021Jhonatan ContrerasPas encore d'évaluation
- Implante SubdérmicoDocument2 pagesImplante SubdérmicoNatali AguileraPas encore d'évaluation
- Gingivitis Descamativa.Document3 pagesGingivitis Descamativa.Andrea QuevedoPas encore d'évaluation
- Caso N° 02Document6 pagesCaso N° 02Fátima SolanshPas encore d'évaluation
- Semiología Del Sistema NerviosoDocument59 pagesSemiología Del Sistema NerviosoSteve Médico100% (1)
- Revisión SíncopeDocument9 pagesRevisión Síncopemedicina_intrerna1Pas encore d'évaluation
- 09 - Guía Práctica Semana 9Document76 pages09 - Guía Práctica Semana 9Andrés MesíaPas encore d'évaluation
- TTo Bio CrohnDocument26 pagesTTo Bio CrohnRolando Illescas QuirozPas encore d'évaluation
- Plan de Cuidados de HidrocefaliaDocument5 pagesPlan de Cuidados de HidrocefaliaLucero Meléndez Pretel100% (1)
- Caso Clínico Practica 1Document3 pagesCaso Clínico Practica 1Nexides RiveraPas encore d'évaluation
- Tipos de AbortosDocument5 pagesTipos de AbortosSthephen CurryPas encore d'évaluation
- Segunda Entrega - LógicaDocument2 pagesSegunda Entrega - LógicaRedAntPas encore d'évaluation
- Enfermedad de Addison (Autoguardado)Document16 pagesEnfermedad de Addison (Autoguardado)Anonymous Hdwjp94k8Pas encore d'évaluation
- Síndrome de Muerte SúbitaDocument12 pagesSíndrome de Muerte Súbitalaura rosalesPas encore d'évaluation
- Cuestionario CardiologíaDocument8 pagesCuestionario CardiologíaLander Duberly Mejia ChullePas encore d'évaluation
- Norma MINSAL ClozapinaDocument37 pagesNorma MINSAL ClozapinaAlan Lama Suazo100% (1)
- Soy Ide Cristian PonceDocument15 pagesSoy Ide Cristian PonceFreddy PanchiPas encore d'évaluation
- 02 Historia Medica Del AlumnoDocument2 pages02 Historia Medica Del AlumnoJessica Tagle BallesterosPas encore d'évaluation
- Psicopatologia 1a Edicion EXPO 1Document4 pagesPsicopatologia 1a Edicion EXPO 1Fatima ArmasPas encore d'évaluation
- Infecciones Vaginales Emilio ChavezDocument16 pagesInfecciones Vaginales Emilio ChavezEmilio Yannick Chavez ZamoraPas encore d'évaluation
- Fisiopatología Cardiovascular INSUFICIENCIA CRADIACA CLASE23-10Document97 pagesFisiopatología Cardiovascular INSUFICIENCIA CRADIACA CLASE23-10Luis Mauricio Tapia QuevedoPas encore d'évaluation
- Objetivo 5. IctericiaDocument4 pagesObjetivo 5. Ictericiadanimora20% (1)
- VIHDocument2 pagesVIHALINA TIMOSHKINA ANATOLIEVNAPas encore d'évaluation
- Farmacología Clínica. Semana 8. Orientac Estudio IndependienteDocument6 pagesFarmacología Clínica. Semana 8. Orientac Estudio IndependienteSoli DuranPas encore d'évaluation
- Historia de La Enfermdedad Del SarampionDocument5 pagesHistoria de La Enfermdedad Del Sarampionderli gutierrezPas encore d'évaluation
- Litiasis Renal Apuntes DRDocument2 pagesLitiasis Renal Apuntes DRdanna boniventoPas encore d'évaluation
- Nivel Bajo de Azúcar en SangreDocument5 pagesNivel Bajo de Azúcar en SangreJuan Almanza MosquedaPas encore d'évaluation
- Trifolio de Osteogénesis ImperfectaDocument2 pagesTrifolio de Osteogénesis Imperfectaraquel alejandra escobarPas encore d'évaluation
- Picadura de Abeja MonografiaDocument8 pagesPicadura de Abeja MonografiaIvan GrandePas encore d'évaluation