Vous aimerez peut-être aussi
- Kerala PSC Examination Expected Questions - 359 (RENAISSANCE) - Kerala PSC Helper - Kerala PSC Questions and AnswersDocument5 pagesKerala PSC Examination Expected Questions - 359 (RENAISSANCE) - Kerala PSC Helper - Kerala PSC Questions and AnswersSreedevi KrishnakumarPas encore d'évaluation
- Chapter 3 Stem EdxDocument28 pagesChapter 3 Stem EdxSreedevi KrishnakumarPas encore d'évaluation
- Kerala PSC Malayalam General Knowledge Questions and Answers - 169 - Kerala PSC Malayalam GK Questions - , ( - )Document3 pagesKerala PSC Malayalam General Knowledge Questions and Answers - 169 - Kerala PSC Malayalam GK Questions - , ( - )Sreedevi Krishnakumar0% (1)
- Kerala PSC Examination Expected Questions - 325 (Renaissance) - Kerala PSC Helper - Kerala PSC Questions and AnswersDocument3 pagesKerala PSC Examination Expected Questions - 325 (Renaissance) - Kerala PSC Helper - Kerala PSC Questions and AnswersSreedevi Krishnakumar0% (2)
- Kerala PSC Books 2016 EXAM Preparation Material OnlineDocument7 pagesKerala PSC Books 2016 EXAM Preparation Material OnlineSreedevi KrishnakumarPas encore d'évaluation
- Arndt Eistert SynthesisDocument2 pagesArndt Eistert SynthesisSreedevi KrishnakumarPas encore d'évaluation
- Other Research Assistant (Chemistry) : (CATEGORY No. 634/2014)Document8 pagesOther Research Assistant (Chemistry) : (CATEGORY No. 634/2014)Sreedevi KrishnakumarPas encore d'évaluation
- Chemical NomenclatureDocument12 pagesChemical NomenclatureSreedevi KrishnakumarPas encore d'évaluation
- NPTEL Phase II - Chemistry and Biochemistry - Selected Topics in Co-Ordination ChemistryDocument2 pagesNPTEL Phase II - Chemistry and Biochemistry - Selected Topics in Co-Ordination ChemistrySreedevi KrishnakumarPas encore d'évaluation
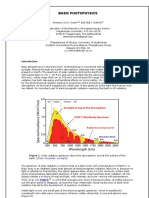
- Basic Photophysics1Document27 pagesBasic Photophysics1Sreedevi KrishnakumarPas encore d'évaluation
- Csir Ugc Net 2008: Chemical Sciences - Paper Ii: Answer Key / Correct Responses OnDocument14 pagesCsir Ugc Net 2008: Chemical Sciences - Paper Ii: Answer Key / Correct Responses OnSreedevi KrishnakumarPas encore d'évaluation
- 5201-MScChem SemI-MQP PDFDocument9 pages5201-MScChem SemI-MQP PDFSreedevi KrishnakumarPas encore d'évaluation
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- The Pendulum KitDocument127 pagesThe Pendulum Kithuman and right89% (9)
- Roots Words of The EnglishDocument2 pagesRoots Words of The EnglishBhagesh Bhosle KhorjuvekarPas encore d'évaluation
- How and Why Did The Art and Visual Culture of The Netherlands Differ From That of Other European Countries During The Seventeenth Century?Document4 pagesHow and Why Did The Art and Visual Culture of The Netherlands Differ From That of Other European Countries During The Seventeenth Century?FranzCondePas encore d'évaluation
- 1 Murid 1 Sukan - ESSEYDocument4 pages1 Murid 1 Sukan - ESSEYUmmuJamilahPas encore d'évaluation
- Hatsumi InterviewDocument2 pagesHatsumi Interviewkosh30700Pas encore d'évaluation
- Basis of Legislation in Islamic State by G A Parwez Published by Idara Tulu-E-IslamDocument12 pagesBasis of Legislation in Islamic State by G A Parwez Published by Idara Tulu-E-IslamadilPas encore d'évaluation
- The Roots of Cognitive Neuroscience - Behavioral Neurology and NeuropsychologyDocument431 pagesThe Roots of Cognitive Neuroscience - Behavioral Neurology and NeuropsychologycharbelhannaPas encore d'évaluation
- FascismDocument71 pagesFascismNitin JainPas encore d'évaluation
- 524 799 Coach - K - Coach - Knight - CaseDocument15 pages524 799 Coach - K - Coach - Knight - Casekaushalmighty100% (1)
- Mahamaya by Rabindranath TagoreDocument1 pageMahamaya by Rabindranath TagoreQuennie100% (7)
- Theories of International Trade and InvestmentDocument35 pagesTheories of International Trade and InvestmentMOHAMMAD ASIF BEGPas encore d'évaluation
- 12 Bal VikasDocument2 pages12 Bal VikasVARUN BPas encore d'évaluation
- Management MKT162Document19 pagesManagement MKT162IzzatSufiIsmailPas encore d'évaluation
- James K.A. Smith On David Foster WallaceDocument3 pagesJames K.A. Smith On David Foster WallaceSancrucensisPas encore d'évaluation
- Cost Optimization ADocument9 pagesCost Optimization AocmainPas encore d'évaluation
- Rubric For Proposal Defense - Project Based: Name: Title: AdviserDocument5 pagesRubric For Proposal Defense - Project Based: Name: Title: AdviserHazel Joy CastilloPas encore d'évaluation
- Kulelat Syndrome 1Document13 pagesKulelat Syndrome 1ava1234567890Pas encore d'évaluation
- Rhetorical AnalysisDocument4 pagesRhetorical Analysisapi-302414270Pas encore d'évaluation
- Berti, Enrico - Ancient Greek Dialectic As Expression of Freedom of Thought and Speech - JHI, 39, 3 - 1978 - 347-370Document25 pagesBerti, Enrico - Ancient Greek Dialectic As Expression of Freedom of Thought and Speech - JHI, 39, 3 - 1978 - 347-370the gatheringPas encore d'évaluation
- Bayessian ClassificationDocument5 pagesBayessian ClassificationManoj BalajiPas encore d'évaluation
- 03 Kesiapsiagaan An MadonaDocument10 pages03 Kesiapsiagaan An MadonaAlamsyah YudhaPas encore d'évaluation
- Narelle McKenzie, Jacqui Showell - Living Fully - Engaging With The Intensity of Life (1998)Document53 pagesNarelle McKenzie, Jacqui Showell - Living Fully - Engaging With The Intensity of Life (1998)Daniel Sun ArguelloPas encore d'évaluation
- Uid 2M QBDocument20 pagesUid 2M QBSuganya NatarajanPas encore d'évaluation
- Ho 5 The Last Castle Traits StyleDocument7 pagesHo 5 The Last Castle Traits StylescarceboyzPas encore d'évaluation
- Take Ving to inf. chưa xảy ra Ving xảy ra rồi: C. not takingDocument5 pagesTake Ving to inf. chưa xảy ra Ving xảy ra rồi: C. not takingTrân GemPas encore d'évaluation
- Becoming An Adaptive Leader PDFDocument8 pagesBecoming An Adaptive Leader PDFPau Sian LianPas encore d'évaluation
- 3.steven Epstein - The Construction of Lay Expertise - AIDS ActivismDocument31 pages3.steven Epstein - The Construction of Lay Expertise - AIDS ActivismTatiana SoaresPas encore d'évaluation
- Condistional SantencesDocument27 pagesCondistional SantencesshizahameedPas encore d'évaluation
- Holt Physics Downloaded Equation SheetDocument2 pagesHolt Physics Downloaded Equation SheetMAXWELL VOGEL50% (2)
- Hodson, Geoffrey - Angels and Archangels (Art)Document3 pagesHodson, Geoffrey - Angels and Archangels (Art)XangotPas encore d'évaluation