Vous aimerez peut-être aussi
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Creative Development of Johann Sebastian Bach, Volume II PDFDocument450 pagesThe Creative Development of Johann Sebastian Bach, Volume II PDFfranelon100% (11)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Homeopatia Vibracional RatesDocument45 pagesHomeopatia Vibracional RatesAugusto Bd100% (4)
- Chrysler Dodge Ram Jeep Drive Cycle InformationDocument2 pagesChrysler Dodge Ram Jeep Drive Cycle InformationslpkthPas encore d'évaluation
- KRPL Shahjahanpur Check List For Arc Welding MachineDocument1 pageKRPL Shahjahanpur Check List For Arc Welding MachineA S YadavPas encore d'évaluation
- The Musical Form Articulates The Traditional Division of The Sonnet Into Four QuatrainsDocument16 pagesThe Musical Form Articulates The Traditional Division of The Sonnet Into Four QuatrainsfranelonPas encore d'évaluation
- A Conductor's Guide To Selected Baroque Choral-OrchestralDocument272 pagesA Conductor's Guide To Selected Baroque Choral-Orchestralfranelon100% (1)
- Collections, Bars and Numbers: Analytical Coincidence or Bach's Design?Document22 pagesCollections, Bars and Numbers: Analytical Coincidence or Bach's Design?franelonPas encore d'évaluation
- Handel's Peace Anthem Author(s) : Donald J. Burrows Source: The Musical Times, Vol. 114, No. 1570 (Dec., 1973), Pp. 1230-1232 Published By: Stable URL: Accessed: 03/08/2014 20:00Document4 pagesHandel's Peace Anthem Author(s) : Donald J. Burrows Source: The Musical Times, Vol. 114, No. 1570 (Dec., 1973), Pp. 1230-1232 Published By: Stable URL: Accessed: 03/08/2014 20:00franelonPas encore d'évaluation
- C.P.E. Bachs Evangelist Johann HeinrichDocument24 pagesC.P.E. Bachs Evangelist Johann HeinrichfranelonPas encore d'évaluation
- Harpsichord in ContinuoDocument11 pagesHarpsichord in ContinuofranelonPas encore d'évaluation
- TotentanzDocument26 pagesTotentanzfranelonPas encore d'évaluation
- Cause List 2.1.2023Document4 pagesCause List 2.1.2023あいうえおかきくけこPas encore d'évaluation
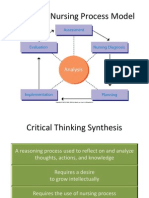
- NUR 104 Nursing Process MY NOTESDocument77 pagesNUR 104 Nursing Process MY NOTESmeanne073100% (1)
- Application Tracking System: Mentor - Yamini Ma'AmDocument10 pagesApplication Tracking System: Mentor - Yamini Ma'AmBHuwanPas encore d'évaluation
- Answer KeyDocument4 pagesAnswer KeyLouina YnciertoPas encore d'évaluation
- Refrigerant Unit Lab ReportDocument19 pagesRefrigerant Unit Lab Reportakmal100% (2)
- Manual Daily Calorie Log: MyfitnesspalDocument4 pagesManual Daily Calorie Log: MyfitnesspalAzariah Burnside100% (2)
- Marisa Wolf Final New ResumeDocument2 pagesMarisa Wolf Final New Resumeapi-403499166Pas encore d'évaluation
- Śāntarak ItaDocument8 pagesŚāntarak ItaÁtilaPas encore d'évaluation
- UVEX - Helmets & Eyewear 2009Document19 pagesUVEX - Helmets & Eyewear 2009Ivica1977Pas encore d'évaluation
- Proac Studio 100: Monitor Level Performance From An Established Compact DesignDocument2 pagesProac Studio 100: Monitor Level Performance From An Established Compact DesignAnonymous c3vuAsWAPas encore d'évaluation
- CIT 811 TMA 4 Quiz QuestionDocument3 pagesCIT 811 TMA 4 Quiz QuestionjohnPas encore d'évaluation
- Electrostatics Practice ProblemsDocument4 pagesElectrostatics Practice ProblemsMohammed Aftab AhmedPas encore d'évaluation
- Cpa f1.1 - Business Mathematics & Quantitative Methods - Study ManualDocument573 pagesCpa f1.1 - Business Mathematics & Quantitative Methods - Study ManualMarcellin MarcaPas encore d'évaluation
- Finite Element Modeling Analysis of Nano Composite Airfoil StructureDocument11 pagesFinite Element Modeling Analysis of Nano Composite Airfoil StructureSuraj GautamPas encore d'évaluation
- British Airways Culture and StructureDocument29 pagesBritish Airways Culture and Structure陆奕敏Pas encore d'évaluation
- Complete DaikinDocument11 pagesComplete DaikinAGNIDEEP BAIDYAPas encore d'évaluation
- Faculty of AyurvedaDocument9 pagesFaculty of AyurvedaKirankumar MutnaliPas encore d'évaluation
- California Academy For Lilminius (Cal) : Lesson PlanDocument4 pagesCalifornia Academy For Lilminius (Cal) : Lesson Plandarryl franciscoPas encore d'évaluation
- 02-Fundamentals of Engineering EconomyDocument14 pages02-Fundamentals of Engineering EconomyLin Xian XingPas encore d'évaluation
- ISA Guidelines PPF 1Document19 pagesISA Guidelines PPF 1Vasu DevanPas encore d'évaluation
- Week 1 Macro (DDR)Document49 pagesWeek 1 Macro (DDR)Stevie Sean100% (1)
- Test Bank Bank For Advanced Accounting 1 E by Bline 382235889 Test Bank Bank For Advanced Accounting 1 E by BlineDocument31 pagesTest Bank Bank For Advanced Accounting 1 E by Bline 382235889 Test Bank Bank For Advanced Accounting 1 E by BlineDe GuzmanPas encore d'évaluation
- FORM Module IpsDocument10 pagesFORM Module IpsRizalPas encore d'évaluation
- HH220i - JAN 11Document1 pageHH220i - JAN 11Achmad GazaliPas encore d'évaluation
- Adolescents and Career DevelopmentDocument10 pagesAdolescents and Career DevelopmentMasrijah MasirPas encore d'évaluation
- The English We SpeakDocument2 pagesThe English We Speakcaeronmustai100% (1)
- 1939 - Hammer - Terrain Corrections For Gravimeter StationsDocument11 pages1939 - Hammer - Terrain Corrections For Gravimeter Stationslinapgeo09100% (1)