Vous aimerez peut-être aussi
- Repro Costanzo NotesDocument11 pagesRepro Costanzo NotesAbeebs SalahouPas encore d'évaluation
- Urinalysis OSCE GuideDocument11 pagesUrinalysis OSCE GuideYu Hsuen Yang0% (1)
- Diseases - BiochemDocument4 pagesDiseases - BiochemJay FeldmanPas encore d'évaluation
- Dokumen - Tips Rubins Pathology Clinicopathologic Foundations of Medicine 7th Edition ChairmanDocument21 pagesDokumen - Tips Rubins Pathology Clinicopathologic Foundations of Medicine 7th Edition ChairmanNare NPas encore d'évaluation
- Ventialtion Perfusion Relationships1Document1 pageVentialtion Perfusion Relationships1DewanggaWahyuPrajaPas encore d'évaluation
- NBME 11 Answers To All Sections 2Document97 pagesNBME 11 Answers To All Sections 2hussainalmusawiPas encore d'évaluation
- Respi PhysioDocument7 pagesRespi PhysioAmal JohnsonPas encore d'évaluation
- Introduction To UrinalysisDocument8 pagesIntroduction To UrinalysisKyle PicocPas encore d'évaluation
- Anat 6.3 GSA Appendix - EsguerraDocument4 pagesAnat 6.3 GSA Appendix - Esguerralovelots1234Pas encore d'évaluation
- OSCE Chart Cough (KK)Document4 pagesOSCE Chart Cough (KK)api-26938624Pas encore d'évaluation
- Anti FungalsDocument5 pagesAnti FungalskakuPas encore d'évaluation
- UW (Step 1) Biochemistry - Educational ObjectivesDocument41 pagesUW (Step 1) Biochemistry - Educational ObjectivesUsama BilalPas encore d'évaluation
- Cocci Rod 4 Main Classifications: Gram Staph, Strep Bacillus Clostridium Neisseria Pleiomorphic Enterobact-EriceaeDocument2 pagesCocci Rod 4 Main Classifications: Gram Staph, Strep Bacillus Clostridium Neisseria Pleiomorphic Enterobact-EriceaeKimberly KanemitsuPas encore d'évaluation
- Histology - Git Table 1Document1 pageHistology - Git Table 1lcrujidoPas encore d'évaluation
- Heart Failure - Notes From "Cardiology" (Timmis Et Al) : Main CausesDocument3 pagesHeart Failure - Notes From "Cardiology" (Timmis Et Al) : Main CausesPrarthana Thiagarajan100% (3)
- Diuretic eDocument2 pagesDiuretic ejustme_adryPas encore d'évaluation
- Zanki (Complete) + R/medicalschoolanki Microbiology ErrataDocument70 pagesZanki (Complete) + R/medicalschoolanki Microbiology ErrataedPas encore d'évaluation
- 7sgdfgf PDFDocument438 pages7sgdfgf PDFPratik JadhavPas encore d'évaluation
- Complete Genetics Disease ChartDocument14 pagesComplete Genetics Disease ChartJames FlanneryPas encore d'évaluation
- Diseases and Deficiencies - USMLE / COMLEXDocument15 pagesDiseases and Deficiencies - USMLE / COMLEXtPas encore d'évaluation
- Actinic KeratosisDocument19 pagesActinic Keratosisattydoc1234Pas encore d'évaluation
- My Notes For USMLEDocument48 pagesMy Notes For USMLEaesfafafaPas encore d'évaluation
- Pediatric Pathology: Disease Cause/Risk Factors SymptomsDocument12 pagesPediatric Pathology: Disease Cause/Risk Factors SymptomsherethemindPas encore d'évaluation
- Respiratory-Equations (Adam Hollingworth)Document4 pagesRespiratory-Equations (Adam Hollingworth)PkernPas encore d'évaluation
- Disease Pathognomonic Sign: Muddy Brown CastsDocument1 pageDisease Pathognomonic Sign: Muddy Brown CastsRafey AhmedPas encore d'évaluation
- Clinical SignsDocument26 pagesClinical SignswiraandiniPas encore d'évaluation
- 2 Renal Buzzword ChartDocument6 pages2 Renal Buzzword ChartTyler KingPas encore d'évaluation
- Table of Genetic Disorders: Download A Copy of This Study GuideDocument11 pagesTable of Genetic Disorders: Download A Copy of This Study Guideerica perezPas encore d'évaluation
- ERRATA 2020 First Aid For The USMLE Step 1Document4 pagesERRATA 2020 First Aid For The USMLE Step 1Rakesh TiwaryPas encore d'évaluation
- USMLE Step 1 DrugsDocument36 pagesUSMLE Step 1 DrugscougardiverPas encore d'évaluation
- Exam 1 DiseasesDocument1 pageExam 1 DiseasesSolomon Seth SallforsPas encore d'évaluation
- 45 NOTES To PG (20 Files Merged)Document338 pages45 NOTES To PG (20 Files Merged)Isak ShatikaPas encore d'évaluation
- Behavioral FinalsDocument28 pagesBehavioral FinalsKofiBPas encore d'évaluation
- AtelectasisDocument3 pagesAtelectasisLouis FortunatoPas encore d'évaluation
- Path DumpsDocument44 pagesPath DumpsAndleeb ImranPas encore d'évaluation
- Hematology & Oncology. Anatomy 56Document60 pagesHematology & Oncology. Anatomy 56Heran TeferiPas encore d'évaluation
- Biostats and Epi-UworldDocument3 pagesBiostats and Epi-UworldMikeRSteinPas encore d'évaluation
- 2-Month Usmle Step 1 Sample ScheduleDocument4 pages2-Month Usmle Step 1 Sample ScheduleSuggula Vamsi KrishnaPas encore d'évaluation
- Heart BlocksDocument9 pagesHeart BlocksDrbee10Pas encore d'évaluation
- Poliomyelitis Haemophilus Influenzae Type B VariecellaDocument4 pagesPoliomyelitis Haemophilus Influenzae Type B VariecellaJeanna Chong100% (1)
- Vasculitis - Student Notes Tabulated2Document2 pagesVasculitis - Student Notes Tabulated2Kirstie de LunaPas encore d'évaluation
- Table of 12 Cranials and TractusDocument5 pagesTable of 12 Cranials and TractusjuwitavalenPas encore d'évaluation
- Arterial Blood Gas Workshop Dr. Lanzona 12.06.07: Lala 3C-Med-09 1Document4 pagesArterial Blood Gas Workshop Dr. Lanzona 12.06.07: Lala 3C-Med-09 1pramastutiPas encore d'évaluation
- DNA Viruses: P P P A H H PDocument2 pagesDNA Viruses: P P P A H H PKimberly KanemitsuPas encore d'évaluation
- Epi Cheatsheet PDFDocument4 pagesEpi Cheatsheet PDFDrbee10Pas encore d'évaluation
- Guideline Pneumothorax PDFDocument1 pageGuideline Pneumothorax PDFRhapsody KarnovinandaPas encore d'évaluation
- Brenner and Stevens, Pharmacology 3 © 2010Document5 pagesBrenner and Stevens, Pharmacology 3 © 2010PharAwayPas encore d'évaluation
- Goljan Blue NotesDocument1 pageGoljan Blue NotesCarl NieweldPas encore d'évaluation
- NBME Exam ContentDocument2 pagesNBME Exam ContentEman ElzeftawyPas encore d'évaluation
- Ahmed Samir - Step 1 Experience - 251Document16 pagesAhmed Samir - Step 1 Experience - 251Mohamed LoaiPas encore d'évaluation
- Step1 Review TopicsDocument32 pagesStep1 Review TopicsAsif AbidiPas encore d'évaluation
- Tumor Markers: Blood Group AntigenDocument5 pagesTumor Markers: Blood Group AntigenAngela ReyesPas encore d'évaluation
- Workbook of BiochemDocument22 pagesWorkbook of BiochemMedStudent MedStudentPas encore d'évaluation
- UWORLDgood NeuroDocument7 pagesUWORLDgood NeurombarreirPas encore d'évaluation
- 2015 Usmle Review Lecture Histology and Cell Biology I Rhys BrooksDocument25 pages2015 Usmle Review Lecture Histology and Cell Biology I Rhys BrooksRushi ShahPas encore d'évaluation
- USmle Radiology 1Document5 pagesUSmle Radiology 1JamesIwuPas encore d'évaluation
- Pathology of The Nervous System PDFDocument17 pagesPathology of The Nervous System PDFAnaPas encore d'évaluation
- Interim Public Health Operational Guidelines For Amoebiasis: (Entamoeba Histolytica)Document34 pagesInterim Public Health Operational Guidelines For Amoebiasis: (Entamoeba Histolytica)QworldPas encore d'évaluation
- Tuberculosis BCG VaccineDocument16 pagesTuberculosis BCG VaccineQworldPas encore d'évaluation
- 5.4 Risk Factors For MRSA ColonizationDocument8 pages5.4 Risk Factors For MRSA ColonizationQworldPas encore d'évaluation
- ID - 13i3 Identification of Pasteurella Species and Morphologically Similar OrganismsDocument28 pagesID - 13i3 Identification of Pasteurella Species and Morphologically Similar OrganismsQworldPas encore d'évaluation
- ID - 12i3 Identification of Haemophilus Species and The HACEK Group of OrganismsDocument35 pagesID - 12i3 Identification of Haemophilus Species and The HACEK Group of OrganismsQworldPas encore d'évaluation
- 5.1 Hand Hygiene Intervention Efficacy ComparisonDocument14 pages5.1 Hand Hygiene Intervention Efficacy ComparisonQworldPas encore d'évaluation
- ID - 12i4 Identification of Haemophilus Species and The HACEKDocument34 pagesID - 12i4 Identification of Haemophilus Species and The HACEKQworldPas encore d'évaluation
- UK Standards For Microbiology Investigations: Indole TestDocument14 pagesUK Standards For Microbiology Investigations: Indole TestQworldPas encore d'évaluation
- TP - 34i4 Thermonuclease TestDocument15 pagesTP - 34i4 Thermonuclease TestQworldPas encore d'évaluation
- TP - 39i3 Staining ProceduresDocument55 pagesTP - 39i3 Staining ProceduresQworldPas encore d'évaluation
- UK Standards For Microbiology Investigations: Catalase TestDocument14 pagesUK Standards For Microbiology Investigations: Catalase TestQworldPas encore d'évaluation
- TP - 5i4 Bile Solubility TestDocument12 pagesTP - 5i4 Bile Solubility TestQworldPas encore d'évaluation
- RNTCP India - Training Module (Dec - 2010) For Medical OfficersDocument128 pagesRNTCP India - Training Module (Dec - 2010) For Medical OfficersDr.Sagindar100% (1)
- Fats and The BrainDocument8 pagesFats and The BrainRade NovakovicPas encore d'évaluation
- Bio MalariaDocument10 pagesBio MalariaAnonymous 42shXQPSjPas encore d'évaluation
- Practice English ConconantsDocument17 pagesPractice English ConconantsStella Trendafilova0% (1)
- CRF TyphoidDocument2 pagesCRF TyphoidAndria GuialalPas encore d'évaluation
- Edema Paru Pit FM SMGDocument36 pagesEdema Paru Pit FM SMGAnonymous ZrLxxRUr9zPas encore d'évaluation
- Animal Testing Essential To Medical ProgressDocument7 pagesAnimal Testing Essential To Medical ProgressLayalPas encore d'évaluation
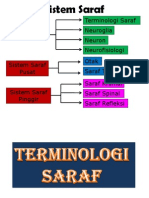
- Tisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafDocument141 pagesTisu Neural Neurofisiologi Neuron Neuroglia Terminologi SarafRainne LeePas encore d'évaluation
- MosquitosDocument2 pagesMosquitosapi-275246146Pas encore d'évaluation
- Soap NoteDocument2 pagesSoap Notetopopirate100% (4)
- Young H Kim Cephalometric Analytic ProcedureDocument13 pagesYoung H Kim Cephalometric Analytic ProcedurePochengShihPas encore d'évaluation
- Protochordata FIXDocument33 pagesProtochordata FIXSylvia AnggraeniPas encore d'évaluation
- AmylaseDocument11 pagesAmylasenestie villaviray100% (1)
- MCQ On Menstrual CycleDocument7 pagesMCQ On Menstrual CycleKarishma Shroff100% (2)
- Ahara VidhiDocument8 pagesAhara VidhishirishkpatilPas encore d'évaluation
- RRLDocument14 pagesRRLFreisanChenMandumotanPas encore d'évaluation
- Cardiac Auscultation - Heart MurmursDocument9 pagesCardiac Auscultation - Heart MurmursAli AftabPas encore d'évaluation
- The Prevalence of Parasitic Protozoan Diseases in Iraq, 2016Document5 pagesThe Prevalence of Parasitic Protozoan Diseases in Iraq, 2016Jeremia AnkesaPas encore d'évaluation
- Bhutan Dream TeerDocument2 pagesBhutan Dream Teerapi-372545986Pas encore d'évaluation
- 10-Bad Moon WaningDocument22 pages10-Bad Moon WaningAllen Zimmerman100% (1)
- Types of Brain WavesDocument1 pageTypes of Brain WavesBluehawk8100% (2)
- Bone HealingDocument2 pagesBone HealingGerardLum100% (2)
- YersiniaDocument46 pagesYersiniajamal nasir50% (2)
- Postnatal Growth and Development: Yenny Yustisia Dept. of Oral Biology Dentistry UNEJDocument38 pagesPostnatal Growth and Development: Yenny Yustisia Dept. of Oral Biology Dentistry UNEJMelisa Novitasari100% (1)
- Final - 2018 CA-1 Tutorial Textbook - Smartphone or Tablet-3Document342 pagesFinal - 2018 CA-1 Tutorial Textbook - Smartphone or Tablet-3Nick-Hugh Wisdom100% (1)
- 20 - Anthrax and BrucellosisDocument4 pages20 - Anthrax and BrucellosisAhmer IsrarPas encore d'évaluation
- ThyroidDocument7 pagesThyroidverawoPas encore d'évaluation
- Block - 3 - 2014 Block 3 BookDocument197 pagesBlock - 3 - 2014 Block 3 BookhappyhappylandPas encore d'évaluation
- Cardiovet OdtDocument26 pagesCardiovet OdtTiberiu CttPas encore d'évaluation
- Ionus Compendium of Strange CreaturesDocument74 pagesIonus Compendium of Strange CreaturesAndrey Augusto100% (3)