Vous aimerez peut-être aussi
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Commonwealth Statutory Declaration Form (May 2011) PDFDocument2 pagesCommonwealth Statutory Declaration Form (May 2011) PDFcgao30Pas encore d'évaluation
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5795)
- Theme IV Yr 5d Content Guide - MatrixDocument9 pagesTheme IV Yr 5d Content Guide - Matrixcgao30Pas encore d'évaluation
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- CPPREP4002 - Annotated Unit GuideDocument8 pagesCPPREP4002 - Annotated Unit Guidecgao30Pas encore d'évaluation
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Pythagoras WorksheetDocument2 pagesPythagoras Worksheetcgao30Pas encore d'évaluation
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- CD Marker Panel Download 2Document2 pagesCD Marker Panel Download 2cgao30Pas encore d'évaluation
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
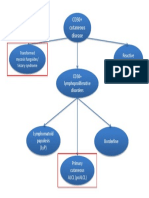
- CD30 Cutaneous Disease FlowchartDocument1 pageCD30 Cutaneous Disease Flowchartcgao30Pas encore d'évaluation
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Paediatrics: Acyanotic Heart DiseaseDocument5 pagesPaediatrics: Acyanotic Heart Diseasecgao30Pas encore d'évaluation
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Matrix TopicsDocument3 pagesMatrix Topicscgao30Pas encore d'évaluation
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Fibroids: 1. Red DegenerationDocument2 pagesFibroids: 1. Red Degenerationcgao30Pas encore d'évaluation
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Pelvic Organ ProlapseDocument5 pagesPelvic Organ Prolapsecgao30Pas encore d'évaluation
- Periodicity NotesDocument5 pagesPeriodicity Notescgao30Pas encore d'évaluation
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- Preterm LabourDocument3 pagesPreterm Labourcgao30Pas encore d'évaluation
- Skin Terms: DescriptorsDocument2 pagesSkin Terms: Descriptorscgao30Pas encore d'évaluation
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Intro To Motor Systems: (Why Else Would You Go To The Gym, Right?)Document13 pagesIntro To Motor Systems: (Why Else Would You Go To The Gym, Right?)cgao30Pas encore d'évaluation
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Pathology Lecture 7 - LiverDocument11 pagesPathology Lecture 7 - Livercgao30Pas encore d'évaluation
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- Kinetics Notes: 6.1 - Rates of ReactionDocument15 pagesKinetics Notes: 6.1 - Rates of Reactioncgao30Pas encore d'évaluation
- Equilibrium NotesDocument5 pagesEquilibrium Notescgao30Pas encore d'évaluation
- Acids and Bases NotesDocument15 pagesAcids and Bases Notescgao30Pas encore d'évaluation
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- Atomic Structure NotesDocument9 pagesAtomic Structure Notescgao30Pas encore d'évaluation
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Atomic Structure NotesDocument9 pagesAtomic Structure Notescgao30Pas encore d'évaluation
- Webquest Infectious DiseaseDocument6 pagesWebquest Infectious Diseaseapi-242716987Pas encore d'évaluation
- Biotech Qns 1Document4 pagesBiotech Qns 1Sirisha Gaja100% (3)
- 2015 Pak SanofiDocument147 pages2015 Pak SanofiUzair Ul GhaniPas encore d'évaluation
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- Malignant Small Round Cell TumorsDocument12 pagesMalignant Small Round Cell TumorsSaptarshi Ghosh100% (1)
- Instant Cloning Kit Manual (For Amplified Products) : Genei GeneiDocument7 pagesInstant Cloning Kit Manual (For Amplified Products) : Genei GeneiHemant KawalkarPas encore d'évaluation
- Lab3 MicrobiologyDocument4 pagesLab3 MicrobiologyRasoulPas encore d'évaluation
- Clark Zhang: Czhang30@jhmi - EduDocument4 pagesClark Zhang: Czhang30@jhmi - Eduapi-278386371Pas encore d'évaluation
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Мелкоклеточная карциномаDocument250 pagesМелкоклеточная карциномаBertramVrellingPas encore d'évaluation
- Journal of Functional Foods: Plantarum K8 On Skin Moisturizing Activity in Human KeratinocyteDocument7 pagesJournal of Functional Foods: Plantarum K8 On Skin Moisturizing Activity in Human KeratinocyteRika LedyPas encore d'évaluation
- Mendel LawDocument4 pagesMendel Lawleon08jayPas encore d'évaluation
- Disadvantages of Evolutionary ClassificationDocument2 pagesDisadvantages of Evolutionary ClassificationAinred KuroakaPas encore d'évaluation
- General and Comparative Endocrinology: Georgia Thomson-Laing, Christine L. Jasoni, P. Mark LokmanDocument9 pagesGeneral and Comparative Endocrinology: Georgia Thomson-Laing, Christine L. Jasoni, P. Mark LokmanharisankarhsPas encore d'évaluation
- Alternative SplicingDocument25 pagesAlternative Splicingtendril123Pas encore d'évaluation
- PhRMA Marketing Brochure Influences On Prescribing FINALDocument12 pagesPhRMA Marketing Brochure Influences On Prescribing FINALJalwaz TihamiPas encore d'évaluation
- DLL On Changes in Chromosome NumberDocument4 pagesDLL On Changes in Chromosome NumberGu Jun Pyo100% (1)
- Human KaryotypeDocument10 pagesHuman Karyotypeprism1702Pas encore d'évaluation
- Principles of Genetics 6th Edition Snustad Test BankDocument15 pagesPrinciples of Genetics 6th Edition Snustad Test BankMelissaLeetioyq100% (18)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Febrauary2020 Chedule For Surgery N Allied 2Document10 pagesFebrauary2020 Chedule For Surgery N Allied 2Saroj YadavPas encore d'évaluation
- Midterm Activity 2 Family Nursing Care PlanDocument4 pagesMidterm Activity 2 Family Nursing Care PlanElly Lazaro100% (1)
- A Geroscience Perspective On Immune Resilience and Infectious Diseases - A Potential Case For MetforminDocument20 pagesA Geroscience Perspective On Immune Resilience and Infectious Diseases - A Potential Case For MetforminAna Raquel Realista Coelho Dos Santos PedrosaPas encore d'évaluation
- IFU FastQ B27 V5 2021 ENDocument10 pagesIFU FastQ B27 V5 2021 ENnbiolab6659Pas encore d'évaluation
- Worksheet: Cellular Respiration & Cell EnergyDocument4 pagesWorksheet: Cellular Respiration & Cell Energyclaryl alexaPas encore d'évaluation
- Fadwa BadranaDocument28 pagesFadwa BadranaDadoPas encore d'évaluation
- Kips Biology PMC TestDocument134 pagesKips Biology PMC TestZahid hussainPas encore d'évaluation
- Lecture 16 Fate Mapping and Gastrulation in Sea UrchinDocument23 pagesLecture 16 Fate Mapping and Gastrulation in Sea UrchinIQra KanwalPas encore d'évaluation
- Cignal Lenti Reporter HandbookDocument28 pagesCignal Lenti Reporter HandbookAnonymous NoI6N0iiPas encore d'évaluation
- Gall Midge Resistance in Rice GenotypesDocument20 pagesGall Midge Resistance in Rice GenotypesGirish ChakraPas encore d'évaluation
- Pharma Cutoff GenDocument7 pagesPharma Cutoff Gennaitik S TPas encore d'évaluation
- Cytogenetics Notes ReviewerDocument11 pagesCytogenetics Notes ReviewerKimPas encore d'évaluation
- Practicals in GeneticsDocument112 pagesPracticals in GeneticscarlosaeserranoPas encore d'évaluation