Vous aimerez peut-être aussi
- Preguntas Ampliación OpticaDocument74 pagesPreguntas Ampliación OpticawaterleliePas encore d'évaluation
- Moises Meza Tarea 2Document19 pagesMoises Meza Tarea 2MOISES MEZA PELUFFOPas encore d'évaluation
- Sol TG 4.1 EdoDocument2 pagesSol TG 4.1 EdoCatalina MaldonadoPas encore d'évaluation
- Cap5Hamilton AlumnosDocument5 pagesCap5Hamilton AlumnosAlbert RaurichPas encore d'évaluation
- Tarea2 - Moises MezaDocument18 pagesTarea2 - Moises MezaMOISES MEZA PELUFFOPas encore d'évaluation
- Ejercicios 1 y 3Document5 pagesEjercicios 1 y 3DEYSI NATALI LUNA BLANCOPas encore d'évaluation
- Transversales 2c.ipynb - Colaboratory PDFDocument10 pagesTransversales 2c.ipynb - Colaboratory PDFSilvio StrajmanPas encore d'évaluation
- Geometría de Información CuánticaDocument10 pagesGeometría de Información CuánticaMiguel MoralesPas encore d'évaluation
- Practica - Clase 2 - Desarrollo - Límites PDFDocument14 pagesPractica - Clase 2 - Desarrollo - Límites PDFDash AcuarioPas encore d'évaluation
- U4. Ecuaciones DiferencialesDocument6 pagesU4. Ecuaciones DiferencialesAna OrtizPas encore d'évaluation
- Teorema de NoetherDocument2 pagesTeorema de NoetherJoePas encore d'évaluation
- PD11Document4 pagesPD11BLADIMIR CESAR PALACIOS PEREZPas encore d'évaluation
- Universidad de Oviedo: Facultad de Ciencias Grado en Matem AticasDocument14 pagesUniversidad de Oviedo: Facultad de Ciencias Grado en Matem AticasNestor AbadPas encore d'évaluation
- S10.s1 - TRANSFORMADA DE LAPLACEDocument34 pagesS10.s1 - TRANSFORMADA DE LAPLACEhananel_forosPas encore d'évaluation
- Solucionario Examen Final 20769 Bloque BDocument6 pagesSolucionario Examen Final 20769 Bloque BYEFRY SARMIENTO CORTEZPas encore d'évaluation
- Parcial 3 Mecánica Cuántica, Gabriel LeónDocument4 pagesParcial 3 Mecánica Cuántica, Gabriel LeónGabriel LeonPas encore d'évaluation
- Ejercicios 2 Ecuaciones Diferenciales Tarea 3Document3 pagesEjercicios 2 Ecuaciones Diferenciales Tarea 3zulayPas encore d'évaluation
- Ejercicio Maxima VerosimilitudDocument2 pagesEjercicio Maxima VerosimilitudDanilo TroncosoPas encore d'évaluation
- Tarea 3.1Document3 pagesTarea 3.1tannia0% (1)
- Informe Investigativo Transformada de Laplace, Transformada Inversa y Ejercicio PrácticoDocument9 pagesInforme Investigativo Transformada de Laplace, Transformada Inversa y Ejercicio PrácticoGilberto BermudezPas encore d'évaluation
- Ejercicio 1 - Luis QuinteroDocument2 pagesEjercicio 1 - Luis QuinteroJose Elias Aranda MendezPas encore d'évaluation
- Poisson Cap 4Document25 pagesPoisson Cap 4Wilder Gonzalez DiazPas encore d'évaluation
- Taller Ecdi MisilDocument5 pagesTaller Ecdi MisilAndrea 1525Pas encore d'évaluation
- Texto Transformada de LaplaceDocument34 pagesTexto Transformada de LaplaceRenato RodriguezPas encore d'évaluation
- Modelos de Inflación y DesempleoDocument9 pagesModelos de Inflación y DesempleoStward Queirolo VillónPas encore d'évaluation
- Solucion PD2Document21 pagesSolucion PD2Jhoan LaldrupPas encore d'évaluation
- Clase 23 SolDocument5 pagesClase 23 SolLuis AntonioPas encore d'évaluation
- Ecuaciones de LagrangeDocument4 pagesEcuaciones de LagrangePaula Andrea Tello PrietoPas encore d'évaluation
- Pauta I1Document4 pagesPauta I1Daniel Vargas ToroPas encore d'évaluation
- Sturm LiouvilleDocument29 pagesSturm LiouvilleJosuè Priego SanabriaPas encore d'évaluation
- El PÉNDULO OSCILANTEDocument4 pagesEl PÉNDULO OSCILANTEIvan GonzalezPas encore d'évaluation
- D.3.01 Screening-Version 2Document24 pagesD.3.01 Screening-Version 2Karim Sobrino AlcasPas encore d'évaluation
- Tarea 5 A La 9 de AlgebraDocument4 pagesTarea 5 A La 9 de AlgebraVianca Anahi Bautista RomeroPas encore d'évaluation
- Tutoría 05Document10 pagesTutoría 05gonzalezchavezgabriel79Pas encore d'évaluation
- Desarrollo Auxiliar 3Document9 pagesDesarrollo Auxiliar 3Juan Luengo MartínPas encore d'évaluation
- Ejercicios Fisica CuanticaDocument12 pagesEjercicios Fisica CuanticaJuan Daniel Marín ArciniegaPas encore d'évaluation
- Límites de Una Función VectorialDocument1 pageLímites de Una Función VectorialAnonymous gfTvfA8Pas encore d'évaluation
- 4 - Fundamentos Matematicos para El Control Automatico de Procesos PDFDocument82 pages4 - Fundamentos Matematicos para El Control Automatico de Procesos PDFBryan S. ReyesPas encore d'évaluation
- Clase 1 LaplaceDocument17 pagesClase 1 LaplaceEliomar RieraPas encore d'évaluation
- Documento Sin TítuloDocument5 pagesDocumento Sin TítuloMARIA FERNANDA EGUILUZ ROBLESPas encore d'évaluation
- Wuolah Free EjerciciosLagrangianoDocument28 pagesWuolah Free EjerciciosLagrangianopepa pigPas encore d'évaluation
- Guía de Ejercicios de Termodinámica 3 (Versión Beta) PDFDocument51 pagesGuía de Ejercicios de Termodinámica 3 (Versión Beta) PDFElio barrios ibarraPas encore d'évaluation
- Ecuaciones DiferencialesDocument3 pagesEcuaciones DiferencialesPaula CasadoPas encore d'évaluation
- Clase 15 T de LaplaceDocument14 pagesClase 15 T de LaplaceCristian NarváezPas encore d'évaluation
- Tarea 2 SoluciónDocument5 pagesTarea 2 SoluciónLuis Enrique Sanchez Mercado :DPas encore d'évaluation
- Capitulo 7Document7 pagesCapitulo 7brujo1997Pas encore d'évaluation
- Formulario ÓPTICADocument15 pagesFormulario ÓPTICAJuan García MenéndezPas encore d'évaluation
- Teoria de TRANSFORMADA DE LAPLACEDocument19 pagesTeoria de TRANSFORMADA DE LAPLACEJoaquÍn SaldañoPas encore d'évaluation
- Examen 3.1 Con Solución Ecuaciones DiferencialesDocument3 pagesExamen 3.1 Con Solución Ecuaciones DiferencialesJaviPas encore d'évaluation
- 2019 - 1 PDFDocument3 pages2019 - 1 PDFJuan Ignacio Acevedo CantillanaPas encore d'évaluation
- La PlaceDocument6 pagesLa PlaceClaudia VargasPas encore d'évaluation
- Resumen3 IIDDocument5 pagesResumen3 IIDLUIS JAVIER GALINDO GALARRETAPas encore d'évaluation
- Capitulo 6 Transformaciones LinealesDocument16 pagesCapitulo 6 Transformaciones LinealesRaul GalindezPas encore d'évaluation
- Ecuaciones Diferenciales-Transformada de LaplaceDocument35 pagesEcuaciones Diferenciales-Transformada de LaplacezurekorlandoPas encore d'évaluation
- Definición de La Transformada de LaplaceDocument5 pagesDefinición de La Transformada de LaplaceNelson Danilo LopezPas encore d'évaluation
- Cap 14 Termo 2Document8 pagesCap 14 Termo 2REBECA ESTER GARCIA MORENOPas encore d'évaluation
- Circuito RLDocument25 pagesCircuito RLRamon Jacobo0% (1)
- Seriespackdepacks ParcialesDocument404 pagesSeriespackdepacks ParcialesJuan CarlosPas encore d'évaluation
- Bol 3Document2 pagesBol 3Paco97Pas encore d'évaluation
- Tema. PerspectivismoDocument2 pagesTema. PerspectivismoPaco97Pas encore d'évaluation
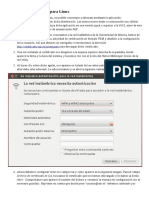
- Eduroam LinuxDocument2 pagesEduroam LinuxPaco97Pas encore d'évaluation
- Historia de La FísicaDocument1 pageHistoria de La FísicaPaco97Pas encore d'évaluation
- Ejercicio 83Document2 pagesEjercicio 83Paco97Pas encore d'évaluation
- Hilbert EquipadosDocument7 pagesHilbert EquipadosPaco97Pas encore d'évaluation
- Foto ElectricoDocument3 pagesFoto ElectricoPaco97Pas encore d'évaluation
- Tarea 1Document2 pagesTarea 1Paco97Pas encore d'évaluation
- t0 PresentacionDocument22 pagest0 PresentacionPaco97Pas encore d'évaluation
- Tarea 7 PenduloDocument2 pagesTarea 7 PenduloPaco97Pas encore d'évaluation
- Calor - Métodos CalorimétricosDocument3 pagesCalor - Métodos CalorimétricosPaco97Pas encore d'évaluation
- BathoryDocument1 pageBathoryPaco97Pas encore d'évaluation
- Introducción A Geometría Diferencial y Teoría de GruposDocument9 pagesIntroducción A Geometría Diferencial y Teoría de GruposPaco97Pas encore d'évaluation
- Sobre La Tesis de EinsteinDocument5 pagesSobre La Tesis de EinsteinPaco97Pas encore d'évaluation
- Péndulo FísicoDocument3 pagesPéndulo FísicoPaco97Pas encore d'évaluation
- Anillos CargadosDocument2 pagesAnillos CargadosPaco97Pas encore d'évaluation
- TermoDocument2 pagesTermoPaco97Pas encore d'évaluation
- Caos DeterministaDocument4 pagesCaos DeterministaPaco97Pas encore d'évaluation
- Circuito RLCDocument4 pagesCircuito RLCPaco97Pas encore d'évaluation
- Péndulo de KáterDocument2 pagesPéndulo de KáterPaco97Pas encore d'évaluation
- Experimento de RutherfordDocument4 pagesExperimento de RutherfordPaco97Pas encore d'évaluation
- QUIMICA-UNIDAD 6 (Soluciones - SolubilidadDocument18 pagesQUIMICA-UNIDAD 6 (Soluciones - SolubilidadAaron AlanizPas encore d'évaluation
- 5 DisolucionesDocument77 pages5 DisolucionesKenia Marcela GonzalezPas encore d'évaluation
- 2020 M&S Clase 3 - Paquete de Fluido - 1parteRevADocument54 pages2020 M&S Clase 3 - Paquete de Fluido - 1parteRevAMaru BaidaPas encore d'évaluation
- Separata de Soluciones o DisolucionesDocument14 pagesSeparata de Soluciones o DisolucionesIng´s-edu.comPas encore d'évaluation
- B1.6-Disoluciones RealesDocument21 pagesB1.6-Disoluciones RealesJoaquin PadillaPas encore d'évaluation
- Reacción 6lk5bvolátilDocument34 pagesReacción 6lk5bvolátilXimena GutierrezPas encore d'évaluation
- Soluciones RealesDocument27 pagesSoluciones RealesbvanoniPas encore d'évaluation
- Exposicion Equilibrio Liquido-VaporDocument27 pagesExposicion Equilibrio Liquido-VaporRomy ArenazasPas encore d'évaluation
- Coeficiente de ActividadDocument11 pagesCoeficiente de ActividadChristopher Castillo GutierrezPas encore d'évaluation
- Soluciones RealesDocument21 pagesSoluciones RealesFer CusimanoPas encore d'évaluation
- 7-Tipos de DestilaciónDocument11 pages7-Tipos de DestilaciónesmeraldaPas encore d'évaluation
- II TemaDocument20 pagesII TemaJuan Manuel Pérez AviñaPas encore d'évaluation
- Práctica 4. Laboratorio de TermodinámicaDocument24 pagesPráctica 4. Laboratorio de TermodinámicaUlises Barrientos SánchezPas encore d'évaluation
- Fisicoquimica II Cohorte 2Document116 pagesFisicoquimica II Cohorte 2Omar Yesid Gomez DurangoPas encore d'évaluation
- Teoría Cinética de Los LíquidosDocument10 pagesTeoría Cinética de Los LíquidosKeila Bermudez100% (1)
- Pauta Auxiliar 03Document13 pagesPauta Auxiliar 03Ignacio Ricardo GomezPas encore d'évaluation
- Previo 4 LqoDocument3 pagesPrevio 4 LqoAbril HerreraPas encore d'évaluation
- TERMOQUIMICA-Termodinamica de Soluciones-FIGMM-UNIDocument31 pagesTERMOQUIMICA-Termodinamica de Soluciones-FIGMM-UNIwalmerPas encore d'évaluation
- 5-1. Termodinámica de Soluciones Poliméricas LDocument67 pages5-1. Termodinámica de Soluciones Poliméricas L17-10636Pas encore d'évaluation
- Manual FisicoquimicaDocument89 pagesManual Fisicoquimicammartinezr26095836Pas encore d'évaluation
- Termodinámica de Las Soluciones (Modo de Compatibilidad) PDFDocument57 pagesTermodinámica de Las Soluciones (Modo de Compatibilidad) PDFjoan davidPas encore d'évaluation
- Informe 2-Mezcla MulticomponentesDocument19 pagesInforme 2-Mezcla MulticomponentesSalustria Cabezas SánchezPas encore d'évaluation
- Torres de AbsorciónDocument10 pagesTorres de AbsorciónAngii KstilloPas encore d'évaluation
- Termo III (Práctica 3)Document33 pagesTermo III (Práctica 3)Jesus Vazquez60% (5)
- 14.en - EsDocument83 pages14.en - EsDiiego CarrilloPas encore d'évaluation
- Disoluciones IdealesDocument10 pagesDisoluciones IdealesCesar Augusto Torres LinaresPas encore d'évaluation
- ElvDocument14 pagesElvNony RechnitzerPas encore d'évaluation
- DISOLUCIONESDocument15 pagesDISOLUCIONESSergio Lazarte Mercado80% (5)
- Ley de RaoultDocument2 pagesLey de RaoultLuxecitaVicentePas encore d'évaluation