Vous aimerez peut-être aussi
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (345)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- PCI Naturals Raw Material Resale Supplier QuestionnaireDocument8 pagesPCI Naturals Raw Material Resale Supplier QuestionnaireJuniorPas encore d'évaluation
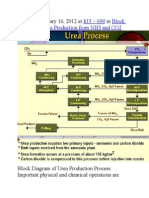
- Published January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2Document9 pagesPublished January 16, 2012 at In: 813 × 699 Block Diagram of Urea Production From NH3 and CO2himanshuchawla654Pas encore d'évaluation
- Reactions of AlkenesDocument37 pagesReactions of Alkenesadamkassas1967Pas encore d'évaluation
- Pyxis SP-910 Portable Procedures Manual PDFDocument286 pagesPyxis SP-910 Portable Procedures Manual PDFKarthikPas encore d'évaluation
- Styrene Based Ion ExchangerDocument35 pagesStyrene Based Ion ExchangerYash PatelPas encore d'évaluation
- Hydrolysis of Esters in Basic SolutionDocument15 pagesHydrolysis of Esters in Basic SolutionZeinab A. ElBhnsawiPas encore d'évaluation
- Dacromet Coating: World Class Corrosion ProtectionDocument6 pagesDacromet Coating: World Class Corrosion Protectionmarcelogf74Pas encore d'évaluation
- Electron Transport ChainDocument2 pagesElectron Transport Chainkshaf muzammil100% (1)
- Biochem Viva Pool (1st Year MBBS)Document11 pagesBiochem Viva Pool (1st Year MBBS)TAHAPas encore d'évaluation
- 1 Emd24Document36 pages1 Emd24Amani KacimiPas encore d'évaluation
- Impact CompoundDocument39 pagesImpact CompoundNabila APas encore d'évaluation
- The Big TEGO. Products Services Data Sheets-75-150-16!76!31-61Document31 pagesThe Big TEGO. Products Services Data Sheets-75-150-16!76!31-61DWI RAHMASARI FATMAWATIPas encore d'évaluation
- Postlab8 9Document3 pagesPostlab8 9Niño Sandro Jocson MercadoPas encore d'évaluation
- As 4351.2-1996 Biodegradability - Organic Compounds in An Aqueous Medium Determination by Analysis of DissolvDocument7 pagesAs 4351.2-1996 Biodegradability - Organic Compounds in An Aqueous Medium Determination by Analysis of DissolvSAI Global - APACPas encore d'évaluation
- Thermal Decomposition of Poly (1,4-Dioxan-2-One)Document12 pagesThermal Decomposition of Poly (1,4-Dioxan-2-One)karinagcarvalhoPas encore d'évaluation
- 2 V 0 ZDocument114 pages2 V 0 ZdulPas encore d'évaluation
- Module 1 - Chemical SafetyDocument21 pagesModule 1 - Chemical SafetyJason ErecillaPas encore d'évaluation
- Product TestingDocument8 pagesProduct TestingmanishrastogiPas encore d'évaluation
- LabDocument4 pagesLabapi-2818620680% (1)
- Fabrication of Hydrophobic and Elastomers Bioplastic Using Polydimethylsiloxane (PDMS)Document12 pagesFabrication of Hydrophobic and Elastomers Bioplastic Using Polydimethylsiloxane (PDMS)ThanhTung NguyenPas encore d'évaluation
- Desmodur N 75 BA/X: Aliphatic PolyisocyanateDocument2 pagesDesmodur N 75 BA/X: Aliphatic PolyisocyanateLê TiếnPas encore d'évaluation
- Paints: Consists of 3 Components: Pigment:-Binder, Vehicle or Resin: - SolventDocument5 pagesPaints: Consists of 3 Components: Pigment:-Binder, Vehicle or Resin: - SolventAshitPas encore d'évaluation
- 8.color Cosmetics-IDocument24 pages8.color Cosmetics-Ikamasuke hegdePas encore d'évaluation
- Plant Alkaloids: Structures and Bioactive PropertiesDocument37 pagesPlant Alkaloids: Structures and Bioactive PropertiesPuskes MYUPas encore d'évaluation
- 9701 w03 Ms 1+2+3+4+5+6Document29 pages9701 w03 Ms 1+2+3+4+5+6Bismaht0% (1)
- The Strategy of Surfactant Analysis by HPLCDocument6 pagesThe Strategy of Surfactant Analysis by HPLCAlex100% (1)
- FarmaVita ManualDocument36 pagesFarmaVita ManualCyanPas encore d'évaluation
- TLC TraduccionDocument16 pagesTLC TraduccionKarlos Lds NvPas encore d'évaluation
- Correlation Soil Properties PDFDocument31 pagesCorrelation Soil Properties PDFBobby SetiawanPas encore d'évaluation
- Case Study On Laguna LakeDocument5 pagesCase Study On Laguna LakeAaron Marius JuliusPas encore d'évaluation