Vous aimerez peut-être aussi
- Huckel TheoryDocument6 pagesHuckel TheorydilatedPas encore d'évaluation
- Exercises Problems Answers Chapter 5Document5 pagesExercises Problems Answers Chapter 5A Sibiescu100% (1)
- Differential Forms on Electromagnetic NetworksD'EverandDifferential Forms on Electromagnetic NetworksÉvaluation : 4 sur 5 étoiles4/5 (1)
- MCQ For DSPDocument38 pagesMCQ For DSPAnonymous JnvCyu85100% (3)
- Line - Cable-Parameter-Calculation-metric MMDocument59 pagesLine - Cable-Parameter-Calculation-metric MMJoseph PoplingerPas encore d'évaluation
- Line Cable Parameter Calculation User ManualDocument59 pagesLine Cable Parameter Calculation User Manualamit77999Pas encore d'évaluation
- Fourier Transforms in NMR, Optical, and Mass Spectrometry: A User's HandbookD'EverandFourier Transforms in NMR, Optical, and Mass Spectrometry: A User's HandbookPas encore d'évaluation
- AP Quantum Numbers WorksheetDocument2 pagesAP Quantum Numbers WorksheetDanah Faith Vera Cruz100% (1)
- Bandas y DOS de GaAsDocument31 pagesBandas y DOS de GaAsFabio JuradoPas encore d'évaluation
- SalidaDocument47 pagesSalidaAriadna Sánchez CastilloPas encore d'évaluation
- MuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelDocument8 pagesMuhammadZahranFadhillah - 1167030050 - Tugas Mandiri KomtelMohammad Zaky AzhariPas encore d'évaluation
- File 1741Document10 pagesFile 1741Renan DanielPas encore d'évaluation
- NPTEL - Assignment - 2 - Phasor EstimationDocument10 pagesNPTEL - Assignment - 2 - Phasor EstimationAKSH0211Pas encore d'évaluation
- Tutorial QuestionsDocument16 pagesTutorial Questionschandadevils100% (1)
- Modal FRA With NastranDocument13 pagesModal FRA With Nastran5colourbalaPas encore d'évaluation
- Problem Set 1Document4 pagesProblem Set 1Rushabh Mehta100% (1)
- Introduction To The Simulation of Signals and Systems: Please Turn The Page OverDocument5 pagesIntroduction To The Simulation of Signals and Systems: Please Turn The Page OverAmanPas encore d'évaluation
- 06 EE 604 1-E - Combine - Combine PDFDocument74 pages06 EE 604 1-E - Combine - Combine PDFdoniya antonyPas encore d'évaluation
- QE Hands-OnDocument9 pagesQE Hands-Onw inxPas encore d'évaluation
- Draft UAS DINAMIKA SISTEM 2022Document6 pagesDraft UAS DINAMIKA SISTEM 2022skiperkresPas encore d'évaluation
- Cruise Control System Using Root Locus MethodDocument8 pagesCruise Control System Using Root Locus Methodsachinsuresh89Pas encore d'évaluation
- FFTDocument28 pagesFFTFan Wang100% (2)
- ADC SNR Performance by Kolsrud PDFDocument37 pagesADC SNR Performance by Kolsrud PDFTuyen DinhPas encore d'évaluation
- Discriminator and Energy Based Demodulators: RevisitedDocument4 pagesDiscriminator and Energy Based Demodulators: RevisitedShakeel RanaPas encore d'évaluation
- Chapter 8 ProblemsDocument21 pagesChapter 8 Problemskhrid3100% (2)
- Be Winter 2020Document2 pagesBe Winter 2020Suraj ThakurPas encore d'évaluation
- 1 Basics DSP AV IntroDocument36 pages1 Basics DSP AV IntroUbaid UmarPas encore d'évaluation
- WIEN2k Getting StartedDocument58 pagesWIEN2k Getting StartedbatistaufrnPas encore d'évaluation
- Bapatla Engineering College::Bapatla: k δ (n−k) - π n) sin Document4 pagesBapatla Engineering College::Bapatla: k δ (n−k) - π n) sin KURRA UPENDRA CHOWDARYPas encore d'évaluation
- Orion Center: Computer Simulation: W.B.MoriDocument24 pagesOrion Center: Computer Simulation: W.B.Moridjyoti96Pas encore d'évaluation
- Spring/Summer 2016 Computer-Based Assignment # 4 Introduction To The Simulation of Signals and SystemsDocument5 pagesSpring/Summer 2016 Computer-Based Assignment # 4 Introduction To The Simulation of Signals and SystemsSelemon RetaPas encore d'évaluation
- Digital Signal Processing (ECE 2203) PDFDocument2 pagesDigital Signal Processing (ECE 2203) PDFAmit ChorariaPas encore d'évaluation
- Obtain The Cascade and Parallel Form Realizations For The Following Systems) Y (N) - 0.1 (n-1) + 0.2 y (n-2) + 3x (N) +3.6 X (n-1) +0.6 X (n-2)Document2 pagesObtain The Cascade and Parallel Form Realizations For The Following Systems) Y (N) - 0.1 (n-1) + 0.2 y (n-2) + 3x (N) +3.6 X (n-1) +0.6 X (n-2)AnnalakshmiPas encore d'évaluation
- National Institute of Technology RourkelaDocument2 pagesNational Institute of Technology RourkelaMrinalPattnaikPas encore d'évaluation
- 2006 ISRO ECE Question Paper PDFDocument19 pages2006 ISRO ECE Question Paper PDFKD0% (1)
- Particle Swarm Optimization (PSO)Document34 pagesParticle Swarm Optimization (PSO)geetesh waghelaPas encore d'évaluation
- DSP ExamDocument2 pagesDSP ExamS. MagidiPas encore d'évaluation
- Digital Signal Processing 2 Sample Midterm Exam: Problem 1: (20 %) Given TheDocument2 pagesDigital Signal Processing 2 Sample Midterm Exam: Problem 1: (20 %) Given TheAn H. TranPas encore d'évaluation
- Experiment No. 2: A. Aim: B. Software To Be Used: C. Equation To Be UsedDocument3 pagesExperiment No. 2: A. Aim: B. Software To Be Used: C. Equation To Be UsedAditya TiwariPas encore d'évaluation
- E. Ambikairajah Australia: Chapter 3 (B) : Digital Signal ProcessingDocument121 pagesE. Ambikairajah Australia: Chapter 3 (B) : Digital Signal ProcessingTahir KhAnPas encore d'évaluation
- 54207-mt - Advanced Digital FilteringDocument2 pages54207-mt - Advanced Digital FilteringSRINIVASA RAO GANTAPas encore d'évaluation
- Grayscale Image Encryption Based On High-Dimensional Fractional Order Chua's SystemDocument2 pagesGrayscale Image Encryption Based On High-Dimensional Fractional Order Chua's SystemPRITAM PARALPas encore d'évaluation
- Lab 2. Generating Cosine and Sine Waves - Real Time DSP Lab ManualDocument5 pagesLab 2. Generating Cosine and Sine Waves - Real Time DSP Lab ManualHamayunPas encore d'évaluation
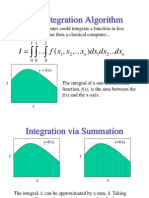
- The Integration Algorithm: DX DX DX X X X F IDocument30 pagesThe Integration Algorithm: DX DX DX X X X F IsuhasnambiarPas encore d'évaluation
- Mat ManualDocument35 pagesMat ManualskandanittePas encore d'évaluation
- Bapatla Engineering College::Bapatla: X (N) 1+e e J JDocument4 pagesBapatla Engineering College::Bapatla: X (N) 1+e e J JKURRA UPENDRA CHOWDARYPas encore d'évaluation
- DPelz IMS2002 WS NBDocument32 pagesDPelz IMS2002 WS NBHüseyin Nuri GülmezPas encore d'évaluation
- DSP Lab RecordDocument37 pagesDSP Lab RecordSathvick BatchuPas encore d'évaluation
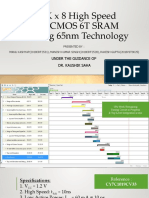
- 128K X 8 High Speed Full CMOS 6T - 4Document32 pages128K X 8 High Speed Full CMOS 6T - 4anwar sayeedPas encore d'évaluation
- II.b.tech-i-sem Bs Lab MDocument190 pagesII.b.tech-i-sem Bs Lab Mshylu ShynyPas encore d'évaluation
- Vlsies Lab Assignment I Sem DSP - Cdac-2Document67 pagesVlsies Lab Assignment I Sem DSP - Cdac-2Jay KothariPas encore d'évaluation
- Ec 2302 DSP EceDocument3 pagesEc 2302 DSP EceIshak Khan GulamPas encore d'évaluation
- University of Technology Department of Electrical Engineering Final Course Examination 2019-2020Document2 pagesUniversity of Technology Department of Electrical Engineering Final Course Examination 2019-2020harithPas encore d'évaluation
- Oldexams DSPDocument25 pagesOldexams DSP29377Pas encore d'évaluation
- GTS User Manual For Finite Element SolverDocument21 pagesGTS User Manual For Finite Element Solverأبو البراءPas encore d'évaluation
- The Fermifab Toolbox For Fermionic Many-Particle Quantum SystemsDocument17 pagesThe Fermifab Toolbox For Fermionic Many-Particle Quantum SystemsAzhar MahmoodPas encore d'évaluation
- Nguyen P Hw2Document7 pagesNguyen P Hw2Phat NguyenPas encore d'évaluation
- Model-Based Processing: An Applied Subspace Identification ApproachD'EverandModel-Based Processing: An Applied Subspace Identification ApproachPas encore d'évaluation
- ZnO SCFDocument1 pageZnO SCFAnugrah Azhar100% (1)
- File: /Media/Azhar/Azhar/A-Nimnsb-Data/Sp Page 1 of 1Document1 pageFile: /Media/Azhar/Azhar/A-Nimnsb-Data/Sp Page 1 of 1Anugrah AzharPas encore d'évaluation
- XM Grace SymbolsDocument5 pagesXM Grace Symbolsbhishan100% (2)
- The Sick LionDocument2 pagesThe Sick LionAnugrah AzharPas encore d'évaluation
- Physics Innovation Project: The Quantum BITDocument6 pagesPhysics Innovation Project: The Quantum BITheywPas encore d'évaluation
- Atomic SanjuDocument42 pagesAtomic Sanjusaptarshi bhattacharyya100% (1)
- Schottky DiodeDocument11 pagesSchottky Diodekkp0650Pas encore d'évaluation
- Potential WellDocument4 pagesPotential WellNaren SundararajanPas encore d'évaluation
- Quantum Entanglement in Fermionic LatticesDocument4 pagesQuantum Entanglement in Fermionic LatticesBrandon ReedPas encore d'évaluation
- Walker4 ISM Ch31Document36 pagesWalker4 ISM Ch31Alejandro Romero MejiaPas encore d'évaluation
- 디지털 공학Document418 pages디지털 공학qkrwlscjf12Pas encore d'évaluation
- Hybridisation For CIEDocument20 pagesHybridisation For CIESyed Umair AnwerPas encore d'évaluation
- Fermi Distribution Function, Effect of Temperature On Fermi FunctionDocument2 pagesFermi Distribution Function, Effect of Temperature On Fermi Functionjainam sharma100% (2)
- Microwave Engineering-Active ComponentsDocument44 pagesMicrowave Engineering-Active ComponentsKobid Karkee100% (2)
- Time Dependent Perturbation ProblemsDocument6 pagesTime Dependent Perturbation ProblemsgitanoganaPas encore d'évaluation
- Progress Report: 1 TM and TM Mixings As Perturbation To Tri-Bimaximal MixingDocument4 pagesProgress Report: 1 TM and TM Mixings As Perturbation To Tri-Bimaximal MixingkanwaljitsinghchanneyPas encore d'évaluation
- Quantum Optics: Marc Torrent CuairanDocument11 pagesQuantum Optics: Marc Torrent CuairanMarc BalaPas encore d'évaluation
- Moon 2019Document13 pagesMoon 2019Vx SimiPas encore d'évaluation
- EEL324: Physical Electronics: Metal-Semiconductor Contacts and Schottky DiodesDocument23 pagesEEL324: Physical Electronics: Metal-Semiconductor Contacts and Schottky DiodesVishal TomarPas encore d'évaluation
- Thin-Film Structuring: Holger BartolfDocument7 pagesThin-Film Structuring: Holger BartolfMichelle CedPas encore d'évaluation
- Quantum Field Theory NotesDocument8 pagesQuantum Field Theory NotesAhmad Afiq bin Ahmad HattaPas encore d'évaluation
- Bloch's Theorem - Definition From AnswersDocument1 pageBloch's Theorem - Definition From AnswersJustice SompoPas encore d'évaluation
- Correlation Between Dislocations and Leakage Current of P-N Diodes On A Free-Standing Gan SubstrateDocument5 pagesCorrelation Between Dislocations and Leakage Current of P-N Diodes On A Free-Standing Gan SubstrateAna-Maria DucuPas encore d'évaluation
- Name: K Sai Bhavana Class: Csit Roll No: 21951A3333 SubjectDocument12 pagesName: K Sai Bhavana Class: Csit Roll No: 21951A3333 SubjectK SAI BHAVANAPas encore d'évaluation
- Unit 9 Semiconductor 2023Document6 pagesUnit 9 Semiconductor 2023spcodrPas encore d'évaluation
- Mynotes NobookDocument554 pagesMynotes Nobookgeopaok165Pas encore d'évaluation
- Symmetry Schemes For The Elementary ParticlesDocument14 pagesSymmetry Schemes For The Elementary ParticlesMani Pillai100% (1)
- Vlsi5 BicmosDocument19 pagesVlsi5 BicmosGaurav MehraPas encore d'évaluation
- Semiconductor PDFDocument9 pagesSemiconductor PDFAnoop JaiswalPas encore d'évaluation
- Eetop - CN E-2Document111 pagesEetop - CN E-2王威棟Pas encore d'évaluation
- Chapter7 Questions IslamDocument46 pagesChapter7 Questions IslamNidhi SisodiaPas encore d'évaluation