Vous aimerez peut-être aussi
- Nomenclature of Heterocyclic CompoundsDocument43 pagesNomenclature of Heterocyclic CompoundsgfdgdghPas encore d'évaluation
- Linear Combination of Atomic Orbitals (LCAO) in Homonuclear Diatomic MoleculesDocument2 pagesLinear Combination of Atomic Orbitals (LCAO) in Homonuclear Diatomic MoleculesSuman DasPas encore d'évaluation
- CHAPTER25 HeterocyclesDocument44 pagesCHAPTER25 HeterocyclesRiyanto WidodoPas encore d'évaluation
- Structure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsD'EverandStructure and Properties of Inorganic Solids: International Series of Monographs in Solid State PhysicsPas encore d'évaluation
- CARBONYL CONDENSATION REACTIONS 2 (10 Mei 2013)Document34 pagesCARBONYL CONDENSATION REACTIONS 2 (10 Mei 2013)Mammy Nya AllyaPas encore d'évaluation
- Named Reactions: 6.1. Aldol CondensationDocument17 pagesNamed Reactions: 6.1. Aldol CondensationNikunja samalPas encore d'évaluation
- Module8 PDFDocument40 pagesModule8 PDFFaizan AhmadPas encore d'évaluation
- SCH 402 Nomenclature of Fused Heterocycles PDFDocument17 pagesSCH 402 Nomenclature of Fused Heterocycles PDFSabz PariPas encore d'évaluation
- Stereochemistry Basic Concepts Useful Notes For StudentsDocument26 pagesStereochemistry Basic Concepts Useful Notes For StudentsReddappaPas encore d'évaluation
- Chapter 15 Parallels Between Main Group and Organometallic ChemistryDocument46 pagesChapter 15 Parallels Between Main Group and Organometallic ChemistryTarang BhatiPas encore d'évaluation
- Clusters and Catenation in P-Block: Allotropes of CarbonDocument15 pagesClusters and Catenation in P-Block: Allotropes of Carbonrajender kumarPas encore d'évaluation
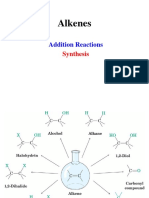
- Alkenes ReactionsDocument69 pagesAlkenes ReactionsAhmad SayyedahmadPas encore d'évaluation
- Resonance and Inductive Effects PresentationDocument36 pagesResonance and Inductive Effects Presentationeagl33yePas encore d'évaluation
- McMurry OC8e EV CH13 PDFDocument28 pagesMcMurry OC8e EV CH13 PDFCrizel Ricaro100% (1)
- TEST 2 GOC & POC Tough by S.K.sinha See Chemistry Animations atDocument3 pagesTEST 2 GOC & POC Tough by S.K.sinha See Chemistry Animations atmyiitchemistryPas encore d'évaluation
- NMR Problems Dec 2012Document8 pagesNMR Problems Dec 2012Biswajit Gopal RoyPas encore d'évaluation
- Carbenes and Nitrenes by IIT CampusDocument13 pagesCarbenes and Nitrenes by IIT CampusHemant soni100% (1)
- H NMR Problems: - How Many Unique Proton Environments Are There inDocument26 pagesH NMR Problems: - How Many Unique Proton Environments Are There inFatima AhmedPas encore d'évaluation
- Reductions in Organic ChemistryDocument13 pagesReductions in Organic ChemistryNikola PudjaPas encore d'évaluation
- Sn1 MechanismDocument24 pagesSn1 MechanismDian MustikasariPas encore d'évaluation
- Chem 212 Alkyl Halide Problems 2Document1 pageChem 212 Alkyl Halide Problems 2kevinamy100% (1)
- Brown 5e Ch07Document33 pagesBrown 5e Ch07Li LizPas encore d'évaluation
- Chem 212 Alkyl Halide Problems 3Document1 pageChem 212 Alkyl Halide Problems 3kevinamyPas encore d'évaluation
- Chemistry 344: Spectroscopy and Spectrometry Problem Set 1Document12 pagesChemistry 344: Spectroscopy and Spectrometry Problem Set 1Fasiha RazaPas encore d'évaluation
- Aromaticity With Huckle's RuleDocument7 pagesAromaticity With Huckle's RuleSk ZPas encore d'évaluation
- Chalcone Synthesis, Structure DiversityDocument13 pagesChalcone Synthesis, Structure DiversityDini Elsi APas encore d'évaluation
- Stereo Chemistry QuestionsDocument19 pagesStereo Chemistry QuestionsfritzPas encore d'évaluation
- Name Reactions-IDocument33 pagesName Reactions-ISatya KamPas encore d'évaluation
- Tutorial 1 @stereochemistry PDFDocument5 pagesTutorial 1 @stereochemistry PDFMoulindu Kundu50% (2)
- IR - HNMR ProblemsDocument33 pagesIR - HNMR Problemsbsakaly112100% (1)
- Azipine PDFDocument58 pagesAzipine PDFGanesamoorthy Thirunarayanan67% (3)
- Comprehensive Test n2 - 2nd Form Sciences - Motherhood Without ArmsDocument4 pagesComprehensive Test n2 - 2nd Form Sciences - Motherhood Without Armsali bettani100% (1)
- Mock Test 5 Paper 1 Q. PaperDocument16 pagesMock Test 5 Paper 1 Q. PaperRare RootPas encore d'évaluation
- Alkyl Halides & Aryl Halides-02 - Solved ProblemsDocument13 pagesAlkyl Halides & Aryl Halides-02 - Solved ProblemsRaju SinghPas encore d'évaluation
- Alcohols & Phenols:: GeneralizationsDocument27 pagesAlcohols & Phenols:: GeneralizationsdoudoudoudouPas encore d'évaluation
- Name Reaction Reagent Assignment PDFDocument21 pagesName Reaction Reagent Assignment PDFSandipan SahaPas encore d'évaluation
- Carbon-Carbon Bond Formation: Comprehensive Organic Synthesis 1991, Vol. 2, 99Document31 pagesCarbon-Carbon Bond Formation: Comprehensive Organic Synthesis 1991, Vol. 2, 99mmiliyasPas encore d'évaluation
- Lab Exercises 1Document3 pagesLab Exercises 1Dinh DungPas encore d'évaluation
- Neighbouring Group ParticipationDocument15 pagesNeighbouring Group ParticipationAbdulMananPas encore d'évaluation
- Silverstein Chapter 1 Mass SpectrometryDocument71 pagesSilverstein Chapter 1 Mass SpectrometryNikita GroverPas encore d'évaluation
- Sigmatropic ReactionDocument14 pagesSigmatropic ReactionAatir HashmiPas encore d'évaluation
- UV-visible Spectrum of A Transition Metal Complex (d3 - d8) From Internet - Assign The Peaks and Using The Appropriate Tanabe - Sugano Diagram Calculate Do For The Complex.Document5 pagesUV-visible Spectrum of A Transition Metal Complex (d3 - d8) From Internet - Assign The Peaks and Using The Appropriate Tanabe - Sugano Diagram Calculate Do For The Complex.peptidesynthesizerPas encore d'évaluation
- Pericyclics-2014 Handout PDFDocument79 pagesPericyclics-2014 Handout PDFnavchemPas encore d'évaluation
- He NeDocument11 pagesHe NeIBN E MARYAMPas encore d'évaluation
- PC11 Molecular Electronic StructureDocument72 pagesPC11 Molecular Electronic StructureAnand MishraPas encore d'évaluation
- EnolateansDocument1 pageEnolateanskevinamyPas encore d'évaluation
- Organic Synthesis. Functional Group InterconversionDocument57 pagesOrganic Synthesis. Functional Group InterconversionJennifer Carolina Rosales NoriegaPas encore d'évaluation
- Electron Delocalization and ResonanceDocument9 pagesElectron Delocalization and ResonanceMariana LizethPas encore d'évaluation
- Mole Concept 2Document38 pagesMole Concept 2R S.NagiPas encore d'évaluation
- Lecture Notes Chapter-12-Aldehydes, Ketones & Carboxylic AcidsDocument26 pagesLecture Notes Chapter-12-Aldehydes, Ketones & Carboxylic AcidsSHUBHAMPas encore d'évaluation
- Hückel's MO Treatment of BenzeneDocument12 pagesHückel's MO Treatment of BenzeneRichard Allen0% (1)
- Haloalkanes PDFDocument6 pagesHaloalkanes PDFthc8477Pas encore d'évaluation
- January 2008 Heterocyclic Chemistry: Exam Questions and Model AnswersDocument17 pagesJanuary 2008 Heterocyclic Chemistry: Exam Questions and Model AnswersPablo de TarsoPas encore d'évaluation
- Aromatic Anti Aromatic Non AromaticDocument2 pagesAromatic Anti Aromatic Non AromaticRitesh SonawanePas encore d'évaluation
- Spectroscopic Solutions of StructureDocument21 pagesSpectroscopic Solutions of StructureKassimPas encore d'évaluation
- Air, Water and Land Pollution: UV-Visible and Infrared Spectroscopic Methods in Environmental AnalysisDocument72 pagesAir, Water and Land Pollution: UV-Visible and Infrared Spectroscopic Methods in Environmental AnalysisSaleem ShaikhPas encore d'évaluation
- Reaction IntermediateDocument20 pagesReaction IntermediateSiddarth Singh100% (2)
- Coordination Compounds - BSC IIIDocument14 pagesCoordination Compounds - BSC IIIRojo John0% (1)
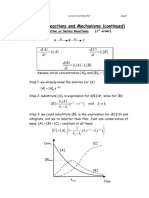
- Consecutive, Series, and Reversible ReactionsDocument6 pagesConsecutive, Series, and Reversible ReactionsRojo JohnPas encore d'évaluation
- Chalk ChromatographyDocument1 pageChalk ChromatographyRojo JohnPas encore d'évaluation
- PH.D Registration Information - 2019 (For Student Supervisor and Place of Research)Document5 pagesPH.D Registration Information - 2019 (For Student Supervisor and Place of Research)Rojo JohnPas encore d'évaluation
- Kinetics of AtmosphereDocument4 pagesKinetics of AtmosphereRojo JohnPas encore d'évaluation
- Two Component Phase Equilibria IDocument6 pagesTwo Component Phase Equilibria IRojo JohnPas encore d'évaluation
- 3 - Rydberg Formula, Photoelectric Effect PDFDocument4 pages3 - Rydberg Formula, Photoelectric Effect PDFRojo JohnPas encore d'évaluation
- Partial Molar PropertiesDocument5 pagesPartial Molar PropertiesRojo JohnPas encore d'évaluation
- 30 - Tanabe Sugano Diagrams PDFDocument5 pages30 - Tanabe Sugano Diagrams PDFRojo JohnPas encore d'évaluation
- Lecture 33: Normal Coordinate AnalysisDocument6 pagesLecture 33: Normal Coordinate AnalysisRojo JohnPas encore d'évaluation
- Multiply Bonded Metal-Ligand ComplexesDocument4 pagesMultiply Bonded Metal-Ligand ComplexesRojo JohnPas encore d'évaluation
- Molecular Point Group 1Document4 pagesMolecular Point Group 1Rojo JohnPas encore d'évaluation
- N-Dimensional Cyclic SystemDocument6 pagesN-Dimensional Cyclic SystemRojo JohnPas encore d'évaluation
- Operator Properties&Mathematical GroupsDocument5 pagesOperator Properties&Mathematical GroupsRojo JohnPas encore d'évaluation
- Liqammonia PDFDocument7 pagesLiqammonia PDFRojo JohnPas encore d'évaluation
- Electronic Selection RulesDocument3 pagesElectronic Selection RulesRojo JohnPas encore d'évaluation
- Spectrochemical SeriesDocument4 pagesSpectrochemical SeriesRojo JohnPas encore d'évaluation
- Stereoselective and Asymmetric Synthesis PDFDocument12 pagesStereoselective and Asymmetric Synthesis PDFRojo JohnPas encore d'évaluation
- D 5917 - 15e1Document8 pagesD 5917 - 15e1Nguyễn Như ThếPas encore d'évaluation
- 2019 Jacs.9b01952 J. Am. Chem. Soc. 2019, 141, 6817 6821 Iridium-Catalyzed Asymmetric Borylation of Unactivated Methylene C (sp3) H BondsDocument5 pages2019 Jacs.9b01952 J. Am. Chem. Soc. 2019, 141, 6817 6821 Iridium-Catalyzed Asymmetric Borylation of Unactivated Methylene C (sp3) H BondsHamza AnsarPas encore d'évaluation
- Carbon Compounds As Fuels & Feedstock 2 QPDocument15 pagesCarbon Compounds As Fuels & Feedstock 2 QPmariamPas encore d'évaluation
- Plant-Based Insect Repellents: A Review of Their Efficacy, Development and TestingDocument16 pagesPlant-Based Insect Repellents: A Review of Their Efficacy, Development and TestingVoulen joy SarenoPas encore d'évaluation
- QUIMICASL Paper3Document43 pagesQUIMICASL Paper3Fiona DonovanPas encore d'évaluation
- Biobutanol: MolassesDocument9 pagesBiobutanol: MolassesElzubair EljaaliPas encore d'évaluation
- Masterbatch Producrers in IndiaDocument39 pagesMasterbatch Producrers in IndiaFuture Innovations100% (1)
- Astm d5453 On-LineDocument11 pagesAstm d5453 On-LineMartua SaragihPas encore d'évaluation
- Proteins SchematicDocument9 pagesProteins SchematicRuchie Ann Pono BaraquilPas encore d'évaluation
- Indigo Dye Derived From Indigofera Tinctoria As Natural Food ColorantDocument6 pagesIndigo Dye Derived From Indigofera Tinctoria As Natural Food Colorantkeexalinee muniandyPas encore d'évaluation
- Icient Treatment of Oil Sands PDocument36 pagesIcient Treatment of Oil Sands PLuqmanPas encore d'évaluation
- PblockDocument13 pagesPblockmandalyuvraj1582Pas encore d'évaluation
- Covalent Bonding QP PDFDocument8 pagesCovalent Bonding QP PDFPichuRangerPas encore d'évaluation
- Gujarat Technological UniversityDocument1 pageGujarat Technological UniversityPatel DhruvilPas encore d'évaluation
- RoHS Declaration of ConformityDocument1 pageRoHS Declaration of ConformityModern DesignsPas encore d'évaluation
- Maize As FodderDocument35 pagesMaize As FodderMessi WorkuPas encore d'évaluation
- Effect of Heating On The Characteristics and Chemical Composition of Selected Frying Oils and FatsDocument6 pagesEffect of Heating On The Characteristics and Chemical Composition of Selected Frying Oils and FatsNiere AdolfoPas encore d'évaluation
- Met-Kleen 155: Innovative Cleaning TechnologyDocument1 pageMet-Kleen 155: Innovative Cleaning TechnologyMiguel BorbaPas encore d'évaluation
- ITW Product Catalog21 PDFDocument1 pageITW Product Catalog21 PDFjohnPas encore d'évaluation
- Coordination Chemistry Reviews: Juan Chen, Wesley R. BrowneDocument21 pagesCoordination Chemistry Reviews: Juan Chen, Wesley R. BrowneSiti ZhaafiraPas encore d'évaluation
- Ion-Exchange Methods and IntercalationDocument4 pagesIon-Exchange Methods and IntercalationHimanshu GuptaPas encore d'évaluation
- Preparation of Benzoic AcidDocument7 pagesPreparation of Benzoic AcidRoberta Piras0% (1)
- A Review On PerfumeryDocument14 pagesA Review On PerfumeryKhoiNguyen1205Pas encore d'évaluation
- Safe - Food - Additives TM Abdel G, JBCR 2015Document37 pagesSafe - Food - Additives TM Abdel G, JBCR 2015Juan Jose Escobar P.Pas encore d'évaluation
- Bomba Maag PiñonesDocument2 pagesBomba Maag Piñonesalexander villanuevaPas encore d'évaluation
- Alkaloids: Carbohydrates and Related CompoundsDocument54 pagesAlkaloids: Carbohydrates and Related CompoundsJohn Kevin TanPas encore d'évaluation
- Foods: A Review of Health-Beneficial Properties of OatsDocument23 pagesFoods: A Review of Health-Beneficial Properties of OatsAbraham Jesús Arzeta RíosPas encore d'évaluation
- The Effect of Fertilizers On Crop Yield, Fruit Quality and Plant Nutrition of Organically Grown STRAWBERRY (Fragaria X Ananassa Duch.)Document11 pagesThe Effect of Fertilizers On Crop Yield, Fruit Quality and Plant Nutrition of Organically Grown STRAWBERRY (Fragaria X Ananassa Duch.)Dinko BećirspahićPas encore d'évaluation
- CH 2 PDFDocument31 pagesCH 2 PDFAbdulbari UshPas encore d'évaluation
- SG Rebong Medical Clinic: Update Product Cost and RevenueDocument4 pagesSG Rebong Medical Clinic: Update Product Cost and RevenueSai BomPas encore d'évaluation
- Handbook of Formulating Dermal Applications: A Definitive Practical GuideD'EverandHandbook of Formulating Dermal Applications: A Definitive Practical GuidePas encore d'évaluation
- Organic Chemistry for Schools: Advanced Level and Senior High SchoolD'EverandOrganic Chemistry for Schools: Advanced Level and Senior High SchoolPas encore d'évaluation
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactD'EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactÉvaluation : 5 sur 5 étoiles5/5 (5)
- Periodic Tales: A Cultural History of the Elements, from Arsenic to ZincD'EverandPeriodic Tales: A Cultural History of the Elements, from Arsenic to ZincÉvaluation : 3.5 sur 5 étoiles3.5/5 (137)
- Is That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeD'EverandIs That a Fact?: Frauds, Quacks, and the Real Science of Everyday LifeÉvaluation : 5 sur 5 étoiles5/5 (4)
- Monkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeD'EverandMonkeys, Myths, and Molecules: Separating Fact from Fiction, and the Science of Everyday LifeÉvaluation : 4 sur 5 étoiles4/5 (1)
- AP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeD'EverandAP® Chemistry Crash Course, For the 2020 Exam, Book + Online: Get a Higher Score in Less TimeÉvaluation : 5 sur 5 étoiles5/5 (1)
- The Periodic Table: A Very Short IntroductionD'EverandThe Periodic Table: A Very Short IntroductionÉvaluation : 4.5 sur 5 étoiles4.5/5 (3)
- Tribology: Friction and Wear of Engineering MaterialsD'EverandTribology: Friction and Wear of Engineering MaterialsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Taste: Surprising Stories and Science About Why Food Tastes GoodD'EverandTaste: Surprising Stories and Science About Why Food Tastes GoodÉvaluation : 3 sur 5 étoiles3/5 (20)
- AP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticeD'EverandAP Chemistry Flashcards, Fourth Edition: Up-to-Date Review and PracticePas encore d'évaluation
- Formulating, Packaging, and Marketing of Natural Cosmetic ProductsD'EverandFormulating, Packaging, and Marketing of Natural Cosmetic ProductsPas encore d'évaluation
- Chemistry for Breakfast: The Amazing Science of Everyday LifeD'EverandChemistry for Breakfast: The Amazing Science of Everyday LifeÉvaluation : 4.5 sur 5 étoiles4.5/5 (90)
- Bioplastics: A Home Inventors HandbookD'EverandBioplastics: A Home Inventors HandbookÉvaluation : 4 sur 5 étoiles4/5 (2)
- Ingredients: A Visual Exploration of 75 Additives & 25 Food ProductsD'EverandIngredients: A Visual Exploration of 75 Additives & 25 Food ProductsÉvaluation : 4 sur 5 étoiles4/5 (1)
- The Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsD'EverandThe Disappearing Spoon: And Other True Tales of Madness, Love, and the History of the World from the Periodic Table of the ElementsÉvaluation : 4 sur 5 étoiles4/5 (146)
- Fundamentals of Chemistry: A Modern IntroductionD'EverandFundamentals of Chemistry: A Modern IntroductionÉvaluation : 5 sur 5 étoiles5/5 (1)
- The Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableD'EverandThe Elements We Live By: How Iron Helps Us Breathe, Potassium Lets Us See, and Other Surprising Superpowers of the Periodic TableÉvaluation : 3.5 sur 5 étoiles3.5/5 (22)
- Water-Based Paint Formulations, Vol. 3D'EverandWater-Based Paint Formulations, Vol. 3Évaluation : 4.5 sur 5 étoiles4.5/5 (6)
- ICH Quality Guidelines: An Implementation GuideD'EverandICH Quality Guidelines: An Implementation GuideAndrew TeasdalePas encore d'évaluation
- The Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactD'EverandThe Nature of Drugs Vol. 1: History, Pharmacology, and Social ImpactÉvaluation : 5 sur 5 étoiles5/5 (1)