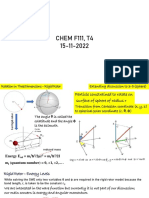
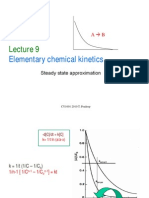
Vous aimerez peut-être aussi
- 6th Central Pay Commission Salary CalculatorDocument15 pages6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Calibration of Pressure GaugeDocument4 pagesCalibration of Pressure GaugeKrish Manglani40% (5)
- 2.01 The Schrodinger Wave Equation For The Hydrogen AtomDocument10 pages2.01 The Schrodinger Wave Equation For The Hydrogen AtomRumbaPas encore d'évaluation
- Muammer Yildiz - Over-Unity Homopolar Electrical Generator - Patent, ArticlesDocument29 pagesMuammer Yildiz - Over-Unity Homopolar Electrical Generator - Patent, ArticlesMohd FakhriPas encore d'évaluation
- Wave Propagation and Power in Lossy DielectricsDocument17 pagesWave Propagation and Power in Lossy DielectricschoiyennPas encore d'évaluation
- BUITEMS Entry Test Sample Paper NAT IMDocument10 pagesBUITEMS Entry Test Sample Paper NAT IMShawn Parker100% (8)
- Practical Physical Pharmaceutics 2012Document33 pagesPractical Physical Pharmaceutics 2012sam100% (2)
- Latticework of Mental Models by Robert HagstromDocument25 pagesLatticework of Mental Models by Robert Hagstromchubbyenoch100% (4)
- B SC Practical Chemistry Gugale PDFDocument316 pagesB SC Practical Chemistry Gugale PDFsam100% (3)
- Identify The Choice That Best Completes The Statement or Answers The QuestionDocument5 pagesIdentify The Choice That Best Completes The Statement or Answers The Questiontwinckel mae bienesPas encore d'évaluation
- Radial Wave Function and Angular Wave FunctionsDocument8 pagesRadial Wave Function and Angular Wave FunctionsBhavesh Garg100% (1)
- Feynman Lectures Simplified 3B: Quantum Mechanics Part TwoD'EverandFeynman Lectures Simplified 3B: Quantum Mechanics Part TwoPas encore d'évaluation
- Radiation Awareness Presentaion (Compatibility Mode)Document40 pagesRadiation Awareness Presentaion (Compatibility Mode)AhmedAmer1Pas encore d'évaluation
- The Kinetic Study of The IodinationDocument6 pagesThe Kinetic Study of The IodinationsamPas encore d'évaluation
- Spectroscopy I (1a)Document6 pagesSpectroscopy I (1a)shaharhr1Pas encore d'évaluation
- Chem5350 Molecular Structure 4upDocument23 pagesChem5350 Molecular Structure 4upUswatul ChasanahPas encore d'évaluation
- Lecturenotes 1Document4 pagesLecturenotes 1Anubhav VardhanPas encore d'évaluation
- Chapter10-Physical Significance of IntegralDocument66 pagesChapter10-Physical Significance of IntegralMika VaughnPas encore d'évaluation
- Postgraduate Physics Course on Molecular SpectroscopyDocument18 pagesPostgraduate Physics Course on Molecular SpectroscopyPragya RawalPas encore d'évaluation
- A HidrogenatomDocument33 pagesA HidrogenatomRévész CsabiPas encore d'évaluation
- Lecture 4 Wavefunction NewDocument53 pagesLecture 4 Wavefunction NewkedirPas encore d'évaluation
- Basics of Atoms, Molecules and Solids: Dr. Rajan Pandey Associate Professor, SENSEDocument19 pagesBasics of Atoms, Molecules and Solids: Dr. Rajan Pandey Associate Professor, SENSEDEEPIKA PAVUNDOSS 20BEC0285Pas encore d'évaluation
- Density Function Theory - PDF 1Document6 pagesDensity Function Theory - PDF 1sanju jarwalPas encore d'évaluation
- Vibrational SpectraDocument27 pagesVibrational SpectraRodrigo Quintana ManfrediPas encore d'évaluation
- Electronic Theory of ChemistryDocument43 pagesElectronic Theory of ChemistryMaheshPas encore d'évaluation
- 01 HydrogenicDocument19 pages01 HydrogenicUlfa WulandariPas encore d'évaluation
- General Chemistry I: Atomic Orbitals and Electron ConfigurationDocument96 pagesGeneral Chemistry I: Atomic Orbitals and Electron ConfigurationKatto - Darling in the PianoPas encore d'évaluation
- Atomic Structure and the Periodic Table in 40 CharactersDocument24 pagesAtomic Structure and the Periodic Table in 40 CharactersRévész CsabiPas encore d'évaluation
- BITS Pilani Chemistry Hamilton OperatorDocument11 pagesBITS Pilani Chemistry Hamilton OperatorNaresh SehdevPas encore d'évaluation
- Unit Ii: Quantum Theory of Hydrogen AtomDocument26 pagesUnit Ii: Quantum Theory of Hydrogen AtomSaqib IrshadPas encore d'évaluation
- Atomic Structure and Atomic SpectraDocument37 pagesAtomic Structure and Atomic SpectraAniSusiloPas encore d'évaluation
- Wave Model - Hydrogen Case - 2023-2024Document36 pagesWave Model - Hydrogen Case - 2023-2024rahmaderradji23Pas encore d'évaluation
- Chapter 8 Atomic StructureDocument68 pagesChapter 8 Atomic StructureHaqnawaz100% (1)
- Lecture 09Document6 pagesLecture 09bgiangre8372Pas encore d'évaluation
- Introduction To Computational Chemistry: Andrew S. IchimuraDocument30 pagesIntroduction To Computational Chemistry: Andrew S. IchimuraAnonymous rn2qoBPjKyPas encore d'évaluation
- Indian Institute of Technology, Guwahati: CH101 Class 11 Physical ChemistryDocument5 pagesIndian Institute of Technology, Guwahati: CH101 Class 11 Physical ChemistryMihir Kumar MechPas encore d'évaluation
- 10 Self-consistent field theory: ² ψ − ∇ ψ dr - ψ - r − r - ψ δ dr ψ - r − r - ψDocument4 pages10 Self-consistent field theory: ² ψ − ∇ ψ dr - ψ - r − r - ψ δ dr ψ - r − r - ψursml12Pas encore d'évaluation
- Harrsion, DFT NatoDocument26 pagesHarrsion, DFT NatosrokkamPas encore d'évaluation
- Lamb Shift Hyperfine Structure Quantum TheoryDocument30 pagesLamb Shift Hyperfine Structure Quantum TheoryBrandon LangleyPas encore d'évaluation
- Approximation Solutions: Multi-E Atoms: RecapDocument10 pagesApproximation Solutions: Multi-E Atoms: RecapDmidPas encore d'évaluation
- Chemistry For EngineersDocument152 pagesChemistry For Engineersmacky 2Pas encore d'évaluation
- CHEM 221/PHY 335 - Molecular Symmetry IDocument40 pagesCHEM 221/PHY 335 - Molecular Symmetry Ipaul javedPas encore d'évaluation
- Atomic structure and the electromagnetic spectrumDocument8 pagesAtomic structure and the electromagnetic spectrumShabnam Fatima Siddiqui100% (1)
- Atomic Structure... (3) PPTDocument32 pagesAtomic Structure... (3) PPTVaibhav KargetiPas encore d'évaluation
- Basics of Lanthanide PhotophysicsDocument45 pagesBasics of Lanthanide PhotophysicsAngela StefanPas encore d'évaluation
- Atomic Units Molecular Hamiltonian Born-Oppenheimer ApproximationDocument7 pagesAtomic Units Molecular Hamiltonian Born-Oppenheimer ApproximationGabriela RadulescuPas encore d'évaluation
- Phonons: 1. Solids in The Adiabatic ApproximationDocument28 pagesPhonons: 1. Solids in The Adiabatic ApproximationcampuspointPas encore d'évaluation
- Quantum Numbers and the Hydrogen AtomDocument26 pagesQuantum Numbers and the Hydrogen AtomWidya FatmawatiPas encore d'évaluation
- II: Experimental Atomic SpectrosDocument21 pagesII: Experimental Atomic SpectrosrogerPas encore d'évaluation
- 2006-7 Module 113 - Lecture 8Document7 pages2006-7 Module 113 - Lecture 8api-19928045Pas encore d'évaluation
- P.F. Bernath - Chapter 16: Electronic Spectroscopy of Diatomic MoleculesDocument13 pagesP.F. Bernath - Chapter 16: Electronic Spectroscopy of Diatomic MoleculesUasnsdaPas encore d'évaluation
- Molecular Reaction DynamicsDocument12 pagesMolecular Reaction DynamicsResourcePas encore d'évaluation
- Module 3 PDFDocument21 pagesModule 3 PDFChintan VasoyaPas encore d'évaluation
- Materi Kimia AnorganikDocument26 pagesMateri Kimia AnorganikNaufal YasinPas encore d'évaluation
- Atomic Physics Modified-RPDocument73 pagesAtomic Physics Modified-RPShrinivas PrabhuPas encore d'évaluation
- Introduction to Computational ChemistryDocument31 pagesIntroduction to Computational ChemistrySonik AlexPas encore d'évaluation
- JChemEduc 1983 60 112-116 PDFDocument5 pagesJChemEduc 1983 60 112-116 PDFLuisPas encore d'évaluation
- Relativistic Effects on Chemical PropertiesDocument5 pagesRelativistic Effects on Chemical PropertiesBeto RodriguezPas encore d'évaluation
- The Schrödinger Wave Equation For The Hydrogen AtomDocument4 pagesThe Schrödinger Wave Equation For The Hydrogen AtomDannie A. San PedroPas encore d'évaluation
- CH2209 - Atomic and Molecular Spectroscopy, Quantum ChemistryDocument18 pagesCH2209 - Atomic and Molecular Spectroscopy, Quantum ChemistryJohnPas encore d'évaluation
- Chemistry Structure of AtomDocument81 pagesChemistry Structure of AtomlololberuhlololPas encore d'évaluation
- Overcoming Limitations of Hartree-Fock for Electron CorrelationDocument7 pagesOvercoming Limitations of Hartree-Fock for Electron CorrelationDiego Alejandro Hurtado BalcazarPas encore d'évaluation
- Atomic Structure: Atomic Structure: Overview of Bohr's Atomic ModelDocument38 pagesAtomic Structure: Atomic Structure: Overview of Bohr's Atomic Modelmanoj kumarPas encore d'évaluation
- From The Schrodinger Equation To Molecular DynamicsDocument16 pagesFrom The Schrodinger Equation To Molecular DynamicsMario Andres JuradoPas encore d'évaluation
- Energy Chem Summary - Libre TextsDocument4 pagesEnergy Chem Summary - Libre Textsmacky 2Pas encore d'évaluation
- Spectroscopic Observation of Helium-Ion-And Hydrogen-Catalyzed Hydrino TransitionsDocument22 pagesSpectroscopic Observation of Helium-Ion-And Hydrogen-Catalyzed Hydrino TransitionsJoão VítorPas encore d'évaluation
- CH 6 (Cont'd)Document5 pagesCH 6 (Cont'd)PineraserPas encore d'évaluation
- Chemistry Unit-2 NotesDocument9 pagesChemistry Unit-2 NotesAkash KumarPas encore d'évaluation
- 14-2 Effects of Electron-Electron Repulsion: 14-3 Exclusion Principle and Exchange InteractionDocument4 pages14-2 Effects of Electron-Electron Repulsion: 14-3 Exclusion Principle and Exchange InteractionFina Aulia RitongaPas encore d'évaluation
- Advances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenD'EverandAdvances in Structure Research by Diffraction Methods: Fortschritte der Strukturforschung mit BeugungsmethodenR. BrillPas encore d'évaluation
- Balungi's Approach to Quantum Gravity: Beyond Einstein, #5D'EverandBalungi's Approach to Quantum Gravity: Beyond Einstein, #5Pas encore d'évaluation
- Zero Order Intergrated Rate Equation PDFDocument1 pageZero Order Intergrated Rate Equation PDFsamPas encore d'évaluation
- Kinetics of The Iodination of Acetone PDFDocument6 pagesKinetics of The Iodination of Acetone PDFsamPas encore d'évaluation
- Integration TypeDocument10 pagesIntegration TypesamPas encore d'évaluation
- Reaction Kinetics ExplainedDocument31 pagesReaction Kinetics ExplainedchweetomahiPas encore d'évaluation
- Chemistry Chapter 1Document3 pagesChemistry Chapter 1faisalPas encore d'évaluation
- Quantum Mechanics, Chapter 8Document7 pagesQuantum Mechanics, Chapter 8oneooninePas encore d'évaluation
- QMChap3 PDFDocument13 pagesQMChap3 PDFsamPas encore d'évaluation
- Bonding Notes5Document25 pagesBonding Notes5Breylon RileyPas encore d'évaluation
- Lecture NoteDocument10 pagesLecture NotesamPas encore d'évaluation
- Notes On Quantum Mechanics: Molecular Structure: Separating Electronic and Nuclear MotionDocument4 pagesNotes On Quantum Mechanics: Molecular Structure: Separating Electronic and Nuclear MotionsamPas encore d'évaluation
- Chemical Kinetics BasicDocument43 pagesChemical Kinetics BasicsamPas encore d'évaluation
- Equi PartitionDocument4 pagesEqui PartitionsamPas encore d'évaluation
- Quantum Mechanics, Chapter 8Document7 pagesQuantum Mechanics, Chapter 8oneooninePas encore d'évaluation
- Water-Phenol Phase DiagramDocument7 pagesWater-Phenol Phase DiagramAkhmad RidhaniPas encore d'évaluation
- Kinetic Theory of Gases and Equipartition TheoremDocument9 pagesKinetic Theory of Gases and Equipartition TheoremsamPas encore d'évaluation
- Bonding Notes5Document25 pagesBonding Notes5Breylon RileyPas encore d'évaluation
- SpectrosDocument5 pagesSpectrossamPas encore d'évaluation
- Lecture 9 Introductory Kinetics PDFDocument78 pagesLecture 9 Introductory Kinetics PDFskrim240Pas encore d'évaluation
- Oxalic AcidDocument2 pagesOxalic Acidsam56% (9)
- Calorimetry ProcedureDocument4 pagesCalorimetry ProceduresamPas encore d'évaluation
- Intro To Black Body RadiationDocument18 pagesIntro To Black Body RadiationtridevmishraPas encore d'évaluation
- Ir SpectrosDocument15 pagesIr SpectrossamPas encore d'évaluation
- Beer's Law CalculationsDocument3 pagesBeer's Law CalculationssamPas encore d'évaluation
- Lecture NoteDocument10 pagesLecture NotesamPas encore d'évaluation
- Bonding Notes5Document25 pagesBonding Notes5Breylon RileyPas encore d'évaluation
- TIFR Physics Previous Year Paper AnalysisDocument3 pagesTIFR Physics Previous Year Paper AnalysisKanishk ChitranshPas encore d'évaluation
- Simulation and Modelling of Water Spray in The 3D ExplosionDocument154 pagesSimulation and Modelling of Water Spray in The 3D Explosionbaal981Pas encore d'évaluation
- Chem 1301 - 2021 MidtermDocument14 pagesChem 1301 - 2021 MidtermRandom PersonPas encore d'évaluation
- Industrial Noise ControlDocument19 pagesIndustrial Noise ControlSantiago IsazaPas encore d'évaluation
- 2023 - Nature - Synthesis-On-substrate of Quantum Dot SolidsDocument18 pages2023 - Nature - Synthesis-On-substrate of Quantum Dot SolidsHridesh GuptaPas encore d'évaluation
- Excel Word Practice Exercise SolutionsDocument9 pagesExcel Word Practice Exercise SolutionsAsif Abdullah KhanPas encore d'évaluation
- Application of Vibration Analysis in The Condition Monitoring of Electrical EquipmentDocument6 pagesApplication of Vibration Analysis in The Condition Monitoring of Electrical EquipmentAlexis AguillonPas encore d'évaluation
- CV BUET (NadimChowdhury) Electrical & Electronic Engineering PDFDocument3 pagesCV BUET (NadimChowdhury) Electrical & Electronic Engineering PDFSujan DebnathPas encore d'évaluation
- Chapter 1-3Document4 pagesChapter 1-3Marvin LiuPas encore d'évaluation
- Homework 07 Solutions: Math 21a Spring, 2014Document6 pagesHomework 07 Solutions: Math 21a Spring, 2014TARANNAUM JAHAN SULTANPas encore d'évaluation
- HIT 2023 Chinese Government Scholarship ProgramDocument32 pagesHIT 2023 Chinese Government Scholarship ProgramSiagalPas encore d'évaluation
- Physics 2 SenguptaDocument38 pagesPhysics 2 SenguptaSashank PrayagaPas encore d'évaluation
- Mechanics ProblemDocument9 pagesMechanics ProblemSon CaoPas encore d'évaluation
- Electro Hydro Forming: Indian Institute of Technology, BhuDocument10 pagesElectro Hydro Forming: Indian Institute of Technology, BhuSourabh KumarPas encore d'évaluation
- 6191 Volume Disk WasherDocument9 pages6191 Volume Disk WasherClifford Ryan GalenzogaPas encore d'évaluation
- (A06) AS Physics (INT) V3aDocument9 pages(A06) AS Physics (INT) V3aharikesh guruPas encore d'évaluation
- Teori Pile Geo5Document27 pagesTeori Pile Geo5Rivana AyuningrumPas encore d'évaluation
- Electromagnetic WavesDocument8 pagesElectromagnetic WavesHafid ZainulPas encore d'évaluation
- Tutorials PTT 205Document2 pagesTutorials PTT 205Celine NathaliaPas encore d'évaluation
- DPP FluidDocument23 pagesDPP FluidAditya SinhaPas encore d'évaluation
- 194 - EE6604 Design of Electrical Machines - Important QuestionsDocument28 pages194 - EE6604 Design of Electrical Machines - Important QuestionsNiteshNarukaPas encore d'évaluation