Vous aimerez peut-être aussi
- Fast Facts: Myelofibrosis: Reviewed by Professor Ruben A. MesaD'EverandFast Facts: Myelofibrosis: Reviewed by Professor Ruben A. MesaPas encore d'évaluation
- Molecularmarkersfor Colorectalcancer: Moriah Wright,, Jenifer S. Beaty,, Charles A. TernentDocument19 pagesMolecularmarkersfor Colorectalcancer: Moriah Wright,, Jenifer S. Beaty,, Charles A. TernentFernando Castro EchavezPas encore d'évaluation
- 10 5772@54652 PDFDocument32 pages10 5772@54652 PDFhollPas encore d'évaluation
- Minimal Residual Disease in Acute Lymphoblastic Leukemia: Dario CampanaDocument6 pagesMinimal Residual Disease in Acute Lymphoblastic Leukemia: Dario CampanaJAIR_valencia3395Pas encore d'évaluation
- 2012 - Intra-Tumour Heterogeneity - A Looking Glass For CancerDocument12 pages2012 - Intra-Tumour Heterogeneity - A Looking Glass For CancerFreddy A. ManayayPas encore d'évaluation
- Tugas DR KamalDocument7 pagesTugas DR KamalZarin SafanahPas encore d'évaluation
- Acute Myeloid Leukemia: Advances in Diagnosis and Classi CationDocument9 pagesAcute Myeloid Leukemia: Advances in Diagnosis and Classi CationTay SalinasPas encore d'évaluation
- 225 VannucchiDocument10 pages225 VannucchiAwais ArshadPas encore d'évaluation
- Chapitre de Livre BoutainaDocument37 pagesChapitre de Livre BoutainaBoutaina AddoumPas encore d'évaluation
- Chronic Lymphocytic Leukemia: A Clinical and Molecular Heterogenous DiseaseDocument14 pagesChronic Lymphocytic Leukemia: A Clinical and Molecular Heterogenous DiseaseCallisthenisLeventisPas encore d'évaluation
- Cancer Total BothDocument33 pagesCancer Total BothUsman AshrafPas encore d'évaluation
- Prognostic Factors in Acute Myeloid Leukaemia 4: Bob LoèwenbergDocument11 pagesPrognostic Factors in Acute Myeloid Leukaemia 4: Bob LoèwenbergStephania SandovalPas encore d'évaluation
- 2020 Genetic Markers in Lung Cancer Diagnosis A ReviewDocument22 pages2020 Genetic Markers in Lung Cancer Diagnosis A Review191933033196Pas encore d'évaluation
- MR 25Document9 pagesMR 25Riko JumattullahPas encore d'évaluation
- Deros A 2015Document10 pagesDeros A 2015rakaPas encore d'évaluation
- Cytogenetic Analysis in The Diagnosis of Acute Leukemia: Sverre Heim, FelixDocument9 pagesCytogenetic Analysis in The Diagnosis of Acute Leukemia: Sverre Heim, FelixEnas KharbotlyPas encore d'évaluation
- Neuroblastoma: Biological Insights Into A Clinical Enigma: Garrett M. BrodeurDocument14 pagesNeuroblastoma: Biological Insights Into A Clinical Enigma: Garrett M. BrodeurMouris DwiputraPas encore d'évaluation
- Ijms 19 03492Document33 pagesIjms 19 03492Qaasim DramatPas encore d'évaluation
- Brock Et Al Liquid Biopsy 2015Document11 pagesBrock Et Al Liquid Biopsy 2015Jean BiverPas encore d'évaluation
- Genetic Alterations and DNA Repair in Human Carcinogenesis: Kathleen Dixon, Elizabeth KoprasDocument8 pagesGenetic Alterations and DNA Repair in Human Carcinogenesis: Kathleen Dixon, Elizabeth Kopras1205lorenaPas encore d'évaluation
- Biomarkers For The Lung Cancer Diagnosis and Their Advances in ProteomicsDocument11 pagesBiomarkers For The Lung Cancer Diagnosis and Their Advances in ProteomicsAndi Harmawati NPas encore d'évaluation
- ADVANCE Molecular Techniques and Histopathology Applications ShortenedDocument5 pagesADVANCE Molecular Techniques and Histopathology Applications ShortenedDale TelgenhoffPas encore d'évaluation
- Chapter 1 - The Cancer GenomeDocument35 pagesChapter 1 - The Cancer GenomeCynthia LopesPas encore d'évaluation
- Unidad VDocument75 pagesUnidad VpablenquePas encore d'évaluation
- Tmp9a87 TMPDocument3 pagesTmp9a87 TMPFrontiersPas encore d'évaluation
- CM7144 Saoirsemorrin 19333910Document51 pagesCM7144 Saoirsemorrin 19333910Saoirse MorrinPas encore d'évaluation
- Plasma Cell Myeloma Literature Review and Case StudyDocument6 pagesPlasma Cell Myeloma Literature Review and Case StudyafdtygyhkPas encore d'évaluation
- Pancreatic CancerDocument13 pagesPancreatic CancerFA MonterPas encore d'évaluation
- Molecular Oncology: Directory of ServicesDocument31 pagesMolecular Oncology: Directory of Servicesruby_kakkar9796100% (2)
- Glioblastoma Stem-Like Cells Approaches For Isolation and CharacterizationDocument19 pagesGlioblastoma Stem-Like Cells Approaches For Isolation and CharacterizationMariano PerezPas encore d'évaluation
- New England Journal Medicine: The ofDocument16 pagesNew England Journal Medicine: The ofMauricio FemeníaPas encore d'évaluation
- Classification of Adult Acute Myeloid LeukemiaDocument24 pagesClassification of Adult Acute Myeloid LeukemiaSusianna RismandaPas encore d'évaluation
- Clinical Implications of BRAF Mutation Test in Colorectal CancerDocument8 pagesClinical Implications of BRAF Mutation Test in Colorectal CancerMohammed AladhraeiPas encore d'évaluation
- Understanding Familial and Non-Familial Renal Cell CancerDocument10 pagesUnderstanding Familial and Non-Familial Renal Cell CancerVanroPas encore d'évaluation
- Molecular Biology As A Tool To Treat CancerDocument8 pagesMolecular Biology As A Tool To Treat CancerFreelance EduPas encore d'évaluation
- Haake Et Al 2017 CancerDocument10 pagesHaake Et Al 2017 Cancerdiego peinadoPas encore d'évaluation
- Giskeødegård Et Al. - Unknown - NMR Based Metabolomics of Biofluids in CancerDocument28 pagesGiskeødegård Et Al. - Unknown - NMR Based Metabolomics of Biofluids in Canceryannick brunatoPas encore d'évaluation
- 1) Explain The Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Document6 pages1) Explain The Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Fahim MahmudPas encore d'évaluation
- Pediatricneuro-Oncology: Fatema MalbariDocument17 pagesPediatricneuro-Oncology: Fatema MalbariRandy UlloaPas encore d'évaluation
- The Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Document7 pagesThe Risk Factors For CLL, Including The Relevance of Monoclonal B Cell Lymphocytosis (MBL)Fahim MahmudPas encore d'évaluation
- Roylance 1997Document8 pagesRoylance 1997barti koksPas encore d'évaluation
- DownloadDocument12 pagesDownloadhasan nazzalPas encore d'évaluation
- JCR 23 007Document13 pagesJCR 23 007yakalapunagarajuPas encore d'évaluation
- Colorectal Cancer PredispositionDocument3 pagesColorectal Cancer PredispositionPete FlintPas encore d'évaluation
- Myeloid Malignancies: Mutations, Models and Management: Review Open AccessDocument15 pagesMyeloid Malignancies: Mutations, Models and Management: Review Open AccessZikry Aulia100% (1)
- 531 (1999) T. R. Golub: Science Et AlDocument8 pages531 (1999) T. R. Golub: Science Et AlBair PuigPas encore d'évaluation
- 1 s2.0 S0304383523000083 MainDocument9 pages1 s2.0 S0304383523000083 MainMericia Guadalupe Sandoval ChavezPas encore d'évaluation
- Tumor ProgressionDocument7 pagesTumor ProgressionmineresearchPas encore d'évaluation
- Cell Cycle and Cancer - Unit I IV/IDocument6 pagesCell Cycle and Cancer - Unit I IV/IManoj GourojuPas encore d'évaluation
- Pi Is 0923753420360798Document16 pagesPi Is 0923753420360798ekanovicaPas encore d'évaluation
- 10 Use of Flow Cytometry in Clonal Plasma Cell DisordersDocument7 pages10 Use of Flow Cytometry in Clonal Plasma Cell DisorderscandiddreamsPas encore d'évaluation
- 5472.can 16 1578Document42 pages5472.can 16 1578肖茹雪Pas encore d'évaluation
- Cancer Res 1990 Wainscoat 1355 60Document7 pagesCancer Res 1990 Wainscoat 1355 60Shahab Ud DinPas encore d'évaluation
- Acute Myeloid Leukemia A Concise ReviewDocument17 pagesAcute Myeloid Leukemia A Concise Reviewberlianza activiraPas encore d'évaluation
- Adult Acute Lymphoblastic Leukemia: Concepts and StrategiesDocument12 pagesAdult Acute Lymphoblastic Leukemia: Concepts and StrategiesdrravesPas encore d'évaluation
- 13 ChemoDocument40 pages13 ChemoSweet SunPas encore d'évaluation
- AlsnlasnfDocument13 pagesAlsnlasnfDina A. ŠabićPas encore d'évaluation
- Acute Promyelocytic Leukemia Part-1Document8 pagesAcute Promyelocytic Leukemia Part-1Francisco Ignacio Reiser ValverdePas encore d'évaluation
- Pages 4 7Document4 pagesPages 4 7andreas_251650Pas encore d'évaluation
- Review Article WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues 4th EditionDocument8 pagesReview Article WHO Classification of Tumors of Hematopoietic and Lymphoid Tissues 4th EditionCat NhanPas encore d'évaluation
- AbemaciclibDocument2 pagesAbemaciclibIna SimachePas encore d'évaluation
- Hallmarks of Cancer An OrganizingDocument4 pagesHallmarks of Cancer An OrganizingIna SimachePas encore d'évaluation
- Alkylating AgentsDocument3 pagesAlkylating AgentsIna SimachePas encore d'évaluation
- Autoimmune Disorders Associated To Type 1Document10 pagesAutoimmune Disorders Associated To Type 1Ina SimachePas encore d'évaluation
- Unified Power Quality Conditioner (Upqc) With Pi and Hysteresis Controller For Power Quality Improvement in Distribution SystemsDocument7 pagesUnified Power Quality Conditioner (Upqc) With Pi and Hysteresis Controller For Power Quality Improvement in Distribution SystemsKANNAN MANIPas encore d'évaluation
- KAHOOT - Assignment 4.1 Lesson PlanDocument3 pagesKAHOOT - Assignment 4.1 Lesson PlanJan ZimmermannPas encore d'évaluation
- 1stQ Week5Document3 pages1stQ Week5Jesse QuingaPas encore d'évaluation
- Hercules Industries Inc. v. Secretary of Labor (1992)Document1 pageHercules Industries Inc. v. Secretary of Labor (1992)Vianca MiguelPas encore d'évaluation
- Julie Jacko - Professor of Healthcare InformaticsDocument1 pageJulie Jacko - Professor of Healthcare InformaticsjuliejackoPas encore d'évaluation
- SEx 3Document33 pagesSEx 3Amir Madani100% (4)
- Windows SCADA Disturbance Capture: User's GuideDocument23 pagesWindows SCADA Disturbance Capture: User's GuideANDREA LILIANA BAUTISTA ACEVEDOPas encore d'évaluation
- Islamic Meditation (Full) PDFDocument10 pagesIslamic Meditation (Full) PDFIslamicfaith Introspection0% (1)
- Lorraln - Corson, Solutions Manual For Electromagnetism - Principles and Applications PDFDocument93 pagesLorraln - Corson, Solutions Manual For Electromagnetism - Principles and Applications PDFc. sorasPas encore d'évaluation
- Barangay Sindalan v. CA G.R. No. 150640, March 22, 2007Document17 pagesBarangay Sindalan v. CA G.R. No. 150640, March 22, 2007FD BalitaPas encore d'évaluation
- Your Free Buyer Persona TemplateDocument8 pagesYour Free Buyer Persona Templateel_nakdjoPas encore d'évaluation
- The Preparedness of The Data Center College of The Philippines To The Flexible Learning Amidst Covid-19 PandemicDocument16 pagesThe Preparedness of The Data Center College of The Philippines To The Flexible Learning Amidst Covid-19 PandemicInternational Journal of Innovative Science and Research TechnologyPas encore d'évaluation
- Accounting 110: Acc110Document19 pagesAccounting 110: Acc110ahoffm05100% (1)
- Hilti Product Technical GuideDocument16 pagesHilti Product Technical Guidegabox707Pas encore d'évaluation
- Kormos - Csizer Language Learning 2008Document29 pagesKormos - Csizer Language Learning 2008Anonymous rDHWR8eBPas encore d'évaluation
- Research ProposalDocument18 pagesResearch ProposalIsmaelPas encore d'évaluation
- Lite Touch. Completo PDFDocument206 pagesLite Touch. Completo PDFkerlystefaniaPas encore d'évaluation
- The Training Toolbox: Forced Reps - The Real Strength SenseiDocument7 pagesThe Training Toolbox: Forced Reps - The Real Strength SenseiSean DrewPas encore d'évaluation
- Name Numerology Calculator - Chaldean Name Number PredictionsDocument2 pagesName Numerology Calculator - Chaldean Name Number Predictionsarunamurugesan7Pas encore d'évaluation
- Focus Group DiscussionDocument13 pagesFocus Group DiscussionSumon ChowdhuryPas encore d'évaluation
- Progressivism Sweeps The NationDocument4 pagesProgressivism Sweeps The NationZach WedelPas encore d'évaluation
- The 5 RS:: A New Teaching Approach To Encourage Slowmations (Student-Generated Animations) of Science ConceptsDocument7 pagesThe 5 RS:: A New Teaching Approach To Encourage Slowmations (Student-Generated Animations) of Science Conceptsnmsharif66Pas encore d'évaluation
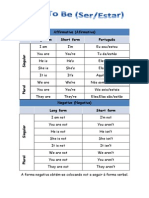
- Affirmative (Afirmativa) Long Form Short Form PortuguêsDocument3 pagesAffirmative (Afirmativa) Long Form Short Form PortuguêsAnitaYangPas encore d'évaluation
- Blunders and How To Avoid Them Dunnington PDFDocument147 pagesBlunders and How To Avoid Them Dunnington PDFrajveer404100% (2)
- Algebra. Equations. Solving Quadratic Equations B PDFDocument1 pageAlgebra. Equations. Solving Quadratic Equations B PDFRoberto CastroPas encore d'évaluation
- Clearing Negative SpiritsDocument6 pagesClearing Negative SpiritsmehorseblessedPas encore d'évaluation
- Nursing Care Plan: Pt.'s Data Nursing Diagnosis GoalsDocument1 pageNursing Care Plan: Pt.'s Data Nursing Diagnosis GoalsKiran Ali100% (3)
- Fansubbers The Case of The Czech Republic and PolandDocument9 pagesFansubbers The Case of The Czech Republic and Polandmusafir24Pas encore d'évaluation
- Leg Res Cases 4Document97 pagesLeg Res Cases 4acheron_pPas encore d'évaluation
- Sino-Japanese Haikai PDFDocument240 pagesSino-Japanese Haikai PDFAlina Diana BratosinPas encore d'évaluation
- 10% Human: How Your Body's Microbes Hold the Key to Health and HappinessD'Everand10% Human: How Your Body's Microbes Hold the Key to Health and HappinessÉvaluation : 4 sur 5 étoiles4/5 (33)
- Masterminds: Genius, DNA, and the Quest to Rewrite LifeD'EverandMasterminds: Genius, DNA, and the Quest to Rewrite LifePas encore d'évaluation
- Why We Die: The New Science of Aging and the Quest for ImmortalityD'EverandWhy We Die: The New Science of Aging and the Quest for ImmortalityÉvaluation : 4.5 sur 5 étoiles4.5/5 (6)
- When the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisD'EverandWhen the Body Says No by Gabor Maté: Key Takeaways, Summary & AnalysisÉvaluation : 3.5 sur 5 étoiles3.5/5 (2)
- Tales from Both Sides of the Brain: A Life in NeuroscienceD'EverandTales from Both Sides of the Brain: A Life in NeuroscienceÉvaluation : 3 sur 5 étoiles3/5 (18)
- The Rise and Fall of the Dinosaurs: A New History of a Lost WorldD'EverandThe Rise and Fall of the Dinosaurs: A New History of a Lost WorldÉvaluation : 4 sur 5 étoiles4/5 (597)
- Buddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomD'EverandBuddha's Brain: The Practical Neuroscience of Happiness, Love & WisdomÉvaluation : 4 sur 5 étoiles4/5 (216)
- Return of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseD'EverandReturn of the God Hypothesis: Three Scientific Discoveries That Reveal the Mind Behind the UniverseÉvaluation : 4.5 sur 5 étoiles4.5/5 (52)
- A Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsD'EverandA Brief History of Intelligence: Evolution, AI, and the Five Breakthroughs That Made Our BrainsÉvaluation : 4.5 sur 5 étoiles4.5/5 (6)
- The Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceD'EverandThe Molecule of More: How a Single Chemical in Your Brain Drives Love, Sex, and Creativity--and Will Determine the Fate of the Human RaceÉvaluation : 4.5 sur 5 étoiles4.5/5 (517)
- Gut: the new and revised Sunday Times bestsellerD'EverandGut: the new and revised Sunday Times bestsellerÉvaluation : 4 sur 5 étoiles4/5 (393)
- Seven and a Half Lessons About the BrainD'EverandSeven and a Half Lessons About the BrainÉvaluation : 4 sur 5 étoiles4/5 (111)
- Undeniable: How Biology Confirms Our Intuition That Life Is DesignedD'EverandUndeniable: How Biology Confirms Our Intuition That Life Is DesignedÉvaluation : 4 sur 5 étoiles4/5 (11)
- All That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesD'EverandAll That Remains: A Renowned Forensic Scientist on Death, Mortality, and Solving CrimesÉvaluation : 4.5 sur 5 étoiles4.5/5 (397)
- The Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionD'EverandThe Ancestor's Tale: A Pilgrimage to the Dawn of EvolutionÉvaluation : 4 sur 5 étoiles4/5 (812)
- Who's in Charge?: Free Will and the Science of the BrainD'EverandWho's in Charge?: Free Will and the Science of the BrainÉvaluation : 4 sur 5 étoiles4/5 (65)
- Good Without God: What a Billion Nonreligious People Do BelieveD'EverandGood Without God: What a Billion Nonreligious People Do BelieveÉvaluation : 4 sur 5 étoiles4/5 (66)
- Moral Tribes: Emotion, Reason, and the Gap Between Us and ThemD'EverandMoral Tribes: Emotion, Reason, and the Gap Between Us and ThemÉvaluation : 4.5 sur 5 étoiles4.5/5 (116)
- Remnants of Ancient Life: The New Science of Old FossilsD'EverandRemnants of Ancient Life: The New Science of Old FossilsÉvaluation : 3 sur 5 étoiles3/5 (3)
- The Lives of Bees: The Untold Story of the Honey Bee in the WildD'EverandThe Lives of Bees: The Untold Story of the Honey Bee in the WildÉvaluation : 4.5 sur 5 étoiles4.5/5 (44)
- Darwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignD'EverandDarwin's Doubt: The Explosive Origin of Animal Life and the Case for Intelligent DesignÉvaluation : 4 sur 5 étoiles4/5 (19)
- The Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindD'EverandThe Consciousness Instinct: Unraveling the Mystery of How the Brain Makes the MindÉvaluation : 4.5 sur 5 étoiles4.5/5 (93)
- Human: The Science Behind What Makes Your Brain UniqueD'EverandHuman: The Science Behind What Makes Your Brain UniqueÉvaluation : 3.5 sur 5 étoiles3.5/5 (38)
- The Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorD'EverandThe Other Side of Normal: How Biology Is Providing the Clues to Unlock the Secrets of Normal and Abnormal BehaviorPas encore d'évaluation