Vous aimerez peut-être aussi
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- 6th Central Pay Commission Salary CalculatorDocument15 pages6th Central Pay Commission Salary Calculatorrakhonde100% (436)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Teaching and Learning StrategiesDocument24 pagesTeaching and Learning Strategiesal alami azzeddine100% (7)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Symmetrical Components 2Document15 pagesSymmetrical Components 2CaribPas encore d'évaluation
- CH 4 Answers PDFDocument26 pagesCH 4 Answers PDFHind Abu GhazlehPas encore d'évaluation
- CH 9 AnswersDocument31 pagesCH 9 AnswersIbrahim A Said100% (2)
- Basics and Statics of Particles - Unit I - GE6253Document57 pagesBasics and Statics of Particles - Unit I - GE6253stkrPas encore d'évaluation
- Activity Sheet Template 1Document8 pagesActivity Sheet Template 1radge edgarPas encore d'évaluation
- SWF - SKmodels - Chain Hoist Operating InstructionsDocument28 pagesSWF - SKmodels - Chain Hoist Operating Instructionsgreek_tester100% (1)
- Medical - History - Questionnaire .Document1 pageMedical - History - Questionnaire .Hind Abu GhazlehPas encore d'évaluation
- Student Progress Sheet Projectile Motion ModelDocument2 pagesStudent Progress Sheet Projectile Motion ModelHind Abu GhazlehPas encore d'évaluation
- Physics SampleDocument9 pagesPhysics SamplevipulmuraliPas encore d'évaluation
- Lesson Plan HindDocument2 pagesLesson Plan HindHind Abu GhazlehPas encore d'évaluation
- Unit 3 - Projectile MotionDocument2 pagesUnit 3 - Projectile MotionHind Abu GhazlehPas encore d'évaluation
- Position Applied For: Direct CV: Candidate BriefDocument1 pagePosition Applied For: Direct CV: Candidate BriefPARVAIZ AHMAD KOKAPas encore d'évaluation
- Unit 3 - Projectile MotionDocument2 pagesUnit 3 - Projectile MotionHind Abu GhazlehPas encore d'évaluation
- Ug PDFDocument265 pagesUg PDFHyakansahsPas encore d'évaluation
- Methanol All DataDocument401 pagesMethanol All DataHind Abu GhazlehPas encore d'évaluation
- Chap 5 Lesson 1Document28 pagesChap 5 Lesson 1Hind Abu GhazlehPas encore d'évaluation
- Lesson Plan HindDocument2 pagesLesson Plan HindHind Abu GhazlehPas encore d'évaluation
- Fortran 1Document44 pagesFortran 1Hind Abu GhazlehPas encore d'évaluation
- 1Document10 pages1Hind Abu GhazlehPas encore d'évaluation
- F 90Document33 pagesF 90Hind Abu GhazlehPas encore d'évaluation
- CH 10 Answers PDFDocument21 pagesCH 10 Answers PDFHind Abu GhazlehPas encore d'évaluation
- A Helping Hand: Author: Payal Dhar Illustrator: Vartika SharmaDocument25 pagesA Helping Hand: Author: Payal Dhar Illustrator: Vartika SharmajedydyPas encore d'évaluation
- I Sing ModelDocument25 pagesI Sing ModelHind Abu GhazlehPas encore d'évaluation
- c3cp52444b 2Document7 pagesc3cp52444b 2Hind Abu GhazlehPas encore d'évaluation
- Jackson 4 13 Homework SolutionDocument3 pagesJackson 4 13 Homework SolutionHind Abu GhazlehPas encore d'évaluation
- Jackson 5 16 Homework SolutionDocument4 pagesJackson 5 16 Homework SolutionHind Abu GhazlehPas encore d'évaluation
- IsingDocument11 pagesIsingHind Abu GhazlehPas encore d'évaluation
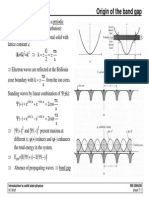
- Origin of the band gap: a x i a x i π − π π − πDocument2 pagesOrigin of the band gap: a x i a x i π − π π − πHind Abu GhazlehPas encore d'évaluation
- Jackson 5 13 Homework SolutionDocument3 pagesJackson 5 13 Homework SolutionHind Abu GhazlehPas encore d'évaluation
- Ising Lenz MCDocument4 pagesIsing Lenz MCHind Abu GhazlehPas encore d'évaluation
- 2.4-Rotational Mechanical System Transfer Functions: Table 3Document5 pages2.4-Rotational Mechanical System Transfer Functions: Table 3طه حسن شاكر محمودPas encore d'évaluation
- 10 Coordination Compound2 (1 Orang)Document11 pages10 Coordination Compound2 (1 Orang)Zakiyah Rizkah Nu'manPas encore d'évaluation
- Degradation Mechanisms of Wire Ropes Operating On Multi-Layer Crane and Mine Hoisting Drums PDFDocument19 pagesDegradation Mechanisms of Wire Ropes Operating On Multi-Layer Crane and Mine Hoisting Drums PDFBruno SantosPas encore d'évaluation
- Media Tension 10Document19 pagesMedia Tension 10EdgardV1024Pas encore d'évaluation
- The Periodic Table and Atomic Theory: Words To KnowDocument10 pagesThe Periodic Table and Atomic Theory: Words To Knowfriscokid13Pas encore d'évaluation
- 2nd Mastery Test ChemistryDocument3 pages2nd Mastery Test Chemistrystephenibahan330Pas encore d'évaluation
- Metal Powder Processing TechniquesDocument26 pagesMetal Powder Processing TechniquesAzhar Ali0% (1)
- Manual Thermo IDocument121 pagesManual Thermo IKevin RamiroPas encore d'évaluation
- SG800GDocument26 pagesSG800GRodrigo CorintoPas encore d'évaluation
- 3382 05a Further Maths - Fp3 Questions v2Document39 pages3382 05a Further Maths - Fp3 Questions v2doctorguy77Pas encore d'évaluation
- Fluid MechanicsDocument2 pagesFluid MechanicsAnkur GoyalPas encore d'évaluation
- Serway 6 e Problems 27Document12 pagesSerway 6 e Problems 27David SaucedoPas encore d'évaluation
- Subatomic ParticleDocument10 pagesSubatomic ParticlemcmusbixPas encore d'évaluation
- tmp91D4 TMPDocument16 pagestmp91D4 TMPFrontiersPas encore d'évaluation
- New Chs PDFDocument28 pagesNew Chs PDFSTUDIESEXAMS ONLYYYPas encore d'évaluation
- Lecture 4 Environmental Instrumental AnalysisDocument68 pagesLecture 4 Environmental Instrumental AnalysisAbu WildanPas encore d'évaluation
- Ijso 2012 MCQDocument15 pagesIjso 2012 MCQmpecth100% (1)
- Welding Shop AssignmentDocument15 pagesWelding Shop AssignmentArslan100% (1)
- Distillation Column T-101 AssignmentDocument30 pagesDistillation Column T-101 AssignmentIlham ZainuddinPas encore d'évaluation
- 06 PlateTheory 09 StrainEnergyDocument4 pages06 PlateTheory 09 StrainEnergyZaid MohammadPas encore d'évaluation
- Atomic Structure: Valence Electrons Determine All of The Following PropertiesDocument7 pagesAtomic Structure: Valence Electrons Determine All of The Following Propertiesjrfr06Pas encore d'évaluation
- Banner SM312DQD DatasheetDocument8 pagesBanner SM312DQD DatasheetemersonmarquezvePas encore d'évaluation
- Chemistry 1F92 Review Final ExamDocument10 pagesChemistry 1F92 Review Final ExamJaredPas encore d'évaluation
- Optical Density and Light SpeedDocument2 pagesOptical Density and Light SpeedLeon MathaiosPas encore d'évaluation
- Chicinas AbsDocument5 pagesChicinas AbsSari Ramadhani MeutuahPas encore d'évaluation
- PolarizationDocument4 pagesPolarizationSwati MishraPas encore d'évaluation