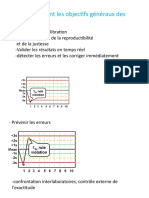
Vous aimerez peut-être aussi
- Chapitre 2 ExtractionDocument19 pagesChapitre 2 ExtractionCHRISTELLE MARINA KOUAKOUPas encore d'évaluation
- 3.propriétés Et TechniquesDocument29 pages3.propriétés Et Techniquessoumia lefkirPas encore d'évaluation
- Extraction ADNDocument19 pagesExtraction ADNkhitriwalid100% (2)
- Cahier D'exercice1Document24 pagesCahier D'exercice1GHERMI .M100% (8)
- Tech Chi ChromatoDocument59 pagesTech Chi ChromatoSamir BENJELLOUN AHMEDPas encore d'évaluation
- La ProtéomiqueDocument36 pagesLa Protéomiqueسلسبيل سلسبيلPas encore d'évaluation
- Cours 3Document40 pagesCours 3Chaima DjabouPas encore d'évaluation
- Extrapolation Des Bioréacteurs (Bioreactor Scale-Up) : Frank DelvigneDocument32 pagesExtrapolation Des Bioréacteurs (Bioreactor Scale-Up) : Frank DelvigneRuben MarquezPas encore d'évaluation
- Strategies en Proteomique Outils Limites Et DeveloDocument10 pagesStrategies en Proteomique Outils Limites Et Develonaomiesolefack03Pas encore d'évaluation
- Seve Michel p17Document18 pagesSeve Michel p17titounanoPas encore d'évaluation
- 1 Proteines 2007-08Document23 pages1 Proteines 2007-08anon_846940093Pas encore d'évaluation
- Biochimie1an-Strategies Etude ProteinesDocument7 pagesBiochimie1an-Strategies Etude ProteinesAGUEZLANE NOURPas encore d'évaluation
- Protéines, Acides Aminés, Enzymes Sériques, ImmunoglobulinesDocument107 pagesProtéines, Acides Aminés, Enzymes Sériques, ImmunoglobulinesTOUIZNI FahimPas encore d'évaluation
- Tech Chi Bio s3 Cours 3Document7 pagesTech Chi Bio s3 Cours 3Ousmane SyllaPas encore d'évaluation
- TD1 Biologie Moléculaire et génie génétiqueDocument25 pagesTD1 Biologie Moléculaire et génie génétiquerosalinarose1995Pas encore d'évaluation
- Cours de Techniques D'analyseDocument70 pagesCours de Techniques D'analyseFaiza Fifi100% (5)
- Cours Chimie Analytique Chapitres 1 Et 2Document10 pagesCours Chimie Analytique Chapitres 1 Et 2Florent yedonouPas encore d'évaluation
- Chapitre 1 - Application Et Analyse Protéomique - 2020Document33 pagesChapitre 1 - Application Et Analyse Protéomique - 2020Mariem YahyaPas encore d'évaluation
- Cours de Toxico Analytique 3 13-4Document4 pagesCours de Toxico Analytique 3 13-4Mebtouche ZianePas encore d'évaluation
- Sujets Concours Magister de Biologie (Biochimie - Microbiologie... )Document32 pagesSujets Concours Magister de Biologie (Biochimie - Microbiologie... )Zineddine Zd33% (3)
- 03-Purification Des Enzymes Et Mesure de Lâ Activitã© EnzymatiqueDocument7 pages03-Purification Des Enzymes Et Mesure de Lâ Activitã© Enzymatique88bkcf9fhq100% (1)
- TP Toxico - OTADocument6 pagesTP Toxico - OTAFouratZarkounaPas encore d'évaluation
- De La Plante A La Formulation QueDocument1 pageDe La Plante A La Formulation Quehakimcab100% (1)
- Application Culture Cellulaire 2020Document28 pagesApplication Culture Cellulaire 2020Manal EzzazaPas encore d'évaluation
- Chapitre 3 InstrumentationsDocument58 pagesChapitre 3 InstrumentationsmjdkjosuePas encore d'évaluation
- Techniques de Chromatogrophie PDFDocument38 pagesTechniques de Chromatogrophie PDFGauthier Djekilamber NendodoumPas encore d'évaluation
- Eléctrophorèse Capillaire APPLICATIONSDocument15 pagesEléctrophorèse Capillaire APPLICATIONSsalim100% (1)
- CIRADjournals,+dk396322Document9 pagesCIRADjournals,+dk396322Etudiant ProPas encore d'évaluation
- SAVE2022 Part 1 ElearnDocument90 pagesSAVE2022 Part 1 ElearnCamilla KafinoPas encore d'évaluation
- TD Fluorescence MoyerDocument5 pagesTD Fluorescence Moyerpriscilla.moyPas encore d'évaluation
- Activite Des Endonucleases de Type II SUR L'ADNDocument26 pagesActivite Des Endonucleases de Type II SUR L'ADNNatacha PoungouPas encore d'évaluation
- Inbound5985577376818810962 PDFDocument34 pagesInbound5985577376818810962 PDFarfaouihekmaPas encore d'évaluation
- Q PCRDocument11 pagesQ PCRBouchra BlsPas encore d'évaluation
- Analyse CouleurDocument11 pagesAnalyse CouleurKarima BakkaliPas encore d'évaluation
- Proteine TechniquesDocument4 pagesProteine TechniquesDonald Njouond KamdemPas encore d'évaluation
- PRABactério001 ECBUDocument4 pagesPRABactério001 ECBUWoppa BahPas encore d'évaluation
- Mémoire de MAGISTER en Biotechnologie: Option: Ecosystèmes Microbiens ComplexesDocument103 pagesMémoire de MAGISTER en Biotechnologie: Option: Ecosystèmes Microbiens ComplexesFayçalPas encore d'évaluation
- Projet de MemoireDocument6 pagesProjet de MemoireBelingaPas encore d'évaluation
- CR TP HPLC - BARRY + TRAORE Binome 8Document20 pagesCR TP HPLC - BARRY + TRAORE Binome 8Manamba Dite Sisse TraoréPas encore d'évaluation
- CH8 Analyses Physicochimiques - ExercicesDocument4 pagesCH8 Analyses Physicochimiques - ExercicesBilal Abou El KacemPas encore d'évaluation
- Chap 5Document27 pagesChap 5Radjaa AddPas encore d'évaluation
- Methode D'etude de Micro-OrganismesDocument9 pagesMethode D'etude de Micro-OrganismesYasser NetPas encore d'évaluation
- Cours de Phylognie MolculaireDocument49 pagesCours de Phylognie MolculaireOrubiyi FADEYIPas encore d'évaluation
- Cours Techniques D'analyses Moléculaires Tabti DDocument71 pagesCours Techniques D'analyses Moléculaires Tabti DHeina JulyPas encore d'évaluation
- Exploration Des Enzymes Sériques Et TissulairesDocument12 pagesExploration Des Enzymes Sériques Et TissulairesmalakPas encore d'évaluation
- TD 1 Toxicologie Analytique-ConvertiDocument41 pagesTD 1 Toxicologie Analytique-ConvertiSelma HassuonPas encore d'évaluation
- ECOTOX MO 16 07 GST v1 2Document10 pagesECOTOX MO 16 07 GST v1 2Alberto MbayoPas encore d'évaluation
- Rapport de Stage 301Document30 pagesRapport de Stage 301Mamadou lamine DiattaPas encore d'évaluation
- Methode de Preconcentration Par Extraction en PhasDocument15 pagesMethode de Preconcentration Par Extraction en PhasJoulia FezzaniPas encore d'évaluation
- Expose de ChromatographieDocument19 pagesExpose de ChromatographieJunior MbatepPas encore d'évaluation
- Protéomique Chapitre 1Document3 pagesProtéomique Chapitre 1belltamer77Pas encore d'évaluation
- Cours - HADDADI GUEMGHAR Hayate - Méthodes SéparativesDocument78 pagesCours - HADDADI GUEMGHAR Hayate - Méthodes SéparativesAl moukhtar BaPas encore d'évaluation
- ProtéomiqueDocument12 pagesProtéomiqueMariem YahyaPas encore d'évaluation
- Cristalographie Des Macromolécules Biologiques 11-11-2014Document45 pagesCristalographie Des Macromolécules Biologiques 11-11-2014Bella Imène100% (1)
- Spectrometrie de Masse MALDI-TOF en BacteriologieDocument7 pagesSpectrometrie de Masse MALDI-TOF en BacteriologieR.Wenceslas LendambaPas encore d'évaluation
- Cours 1 SpectrophotometrieDocument10 pagesCours 1 SpectrophotometriehadilPas encore d'évaluation
- Purification Des ProtéinesDocument69 pagesPurification Des ProtéinesChaima YbPas encore d'évaluation
- La désintoxication douce: Programme de détoxification naturelleD'EverandLa désintoxication douce: Programme de détoxification naturelleÉvaluation : 5 sur 5 étoiles5/5 (1)
- Applications de la spectrophotomérie en phytochimie: sciencesD'EverandApplications de la spectrophotomérie en phytochimie: sciencesPas encore d'évaluation
- Les Protéomes Du FoieDocument41 pagesLes Protéomes Du FoieLamis HamouyPas encore d'évaluation
- These YOUNES SorayaDocument162 pagesThese YOUNES SorayaLamis HamouyPas encore d'évaluation
- CHIKHI - COURS 2 - Introduction - Au - DockingDocument57 pagesCHIKHI - COURS 2 - Introduction - Au - DockingLamis HamouyPas encore d'évaluation
- Les Trois Formes Majeures de Lipides Alimentaires Sont Les TriglycéridesDocument1 pageLes Trois Formes Majeures de Lipides Alimentaires Sont Les TriglycéridesLamis HamouyPas encore d'évaluation
- Annales Médecine Lyon-Nord 2008-2009 PCEM1Document139 pagesAnnales Médecine Lyon-Nord 2008-2009 PCEM1Lamis HamouyPas encore d'évaluation
- Programe LicenceDocument76 pagesPrograme LicenceLamis HamouyPas encore d'évaluation
- 458 Em15102011Document20 pages458 Em15102011elmoudjahid_dzPas encore d'évaluation
- Atelier Ouvrage D'artDocument26 pagesAtelier Ouvrage D'art3 éme infraPas encore d'évaluation
- Cours DeterminantDocument7 pagesCours Determinanttarek gritliPas encore d'évaluation
- SVTDocument3 pagesSVTCheikh DiaPas encore d'évaluation
- QCM La Regulation de La GlycemieDocument4 pagesQCM La Regulation de La GlycemiekamiliaPas encore d'évaluation
- Le Seuil de Rentabilite Exercice CopemDocument2 pagesLe Seuil de Rentabilite Exercice CopemMohammed EL AMRANIPas encore d'évaluation
- Pierre Leyssenne LA PREMIERE ANNEE D'ARITHMETIQUE Librairie Armand Colin Paris 1915Document148 pagesPierre Leyssenne LA PREMIERE ANNEE D'ARITHMETIQUE Librairie Armand Colin Paris 1915francis batt100% (1)
- L'economie de La Cote D'ivoirDocument6 pagesL'economie de La Cote D'ivoirballa pierre koivoguiPas encore d'évaluation
- 2001 Sept PT1Document19 pages2001 Sept PT1HamydPas encore d'évaluation
- Psycho Slides Stat L3 S6Document76 pagesPsycho Slides Stat L3 S6Bvictor Boni100% (1)
- Hematologie GeneraleDocument108 pagesHematologie GeneraleAbdelhedi AmirPas encore d'évaluation
- M. FAYE-TD Cinematique Du PointDocument3 pagesM. FAYE-TD Cinematique Du PointDabo100% (1)
- Pole 1-1Document2 pagesPole 1-1Sabhrya CorbonPas encore d'évaluation
- STARBUCKSDocument18 pagesSTARBUCKSHARMACHEPas encore d'évaluation
- Foulées D'imphy 2019Document5 pagesFoulées D'imphy 2019Le Journal du CentrePas encore d'évaluation
- Umcj Extrait Kryptonite PDFDocument30 pagesUmcj Extrait Kryptonite PDFbalkassangPas encore d'évaluation
- 2ème Devoir Du 2ème Semestre Mathematiques 3ème 2018-2019 Ceg BoucaDocument2 pages2ème Devoir Du 2ème Semestre Mathematiques 3ème 2018-2019 Ceg BoucaPétronePas encore d'évaluation
- Cours D-Astrologie 1-2 PDFDocument8 pagesCours D-Astrologie 1-2 PDFmo50% (2)
- Cours Cycle de Vie 2Document100 pagesCours Cycle de Vie 2slimamriPas encore d'évaluation
- Lecon 1 12Document6 pagesLecon 1 12papillon116Pas encore d'évaluation
- Piarc Revue RoutesDocument68 pagesPiarc Revue RoutesMohamed LouridiPas encore d'évaluation
- Yves Tissier - Etre-Vegetarien - Le-Bon-ChoixDocument245 pagesYves Tissier - Etre-Vegetarien - Le-Bon-ChoixKabiéssi ODOUDEYPas encore d'évaluation
- Mur en Béton ArmeDocument24 pagesMur en Béton ArmenidhalPas encore d'évaluation
- Les ClavettesDocument18 pagesLes Clavetteskhocine100% (1)
- SG EuDocument164 pagesSG EuSander SwinnenPas encore d'évaluation
- Recette Roulés de Jambon Au Beurre de Poireaux GratinésDocument9 pagesRecette Roulés de Jambon Au Beurre de Poireaux GratinésAmina EL HALFIPas encore d'évaluation
- L'anémie RegimeDocument7 pagesL'anémie Regimebendjillali youcefPas encore d'évaluation
- Ensemencement D'une Galarie API 20Document50 pagesEnsemencement D'une Galarie API 20alphonse1988100% (3)
- Documentation Volkswagen New BeetleDocument19 pagesDocumentation Volkswagen New Beetlecristi botezPas encore d'évaluation
- Cataělogo Fassi 175 PDFDocument24 pagesCataělogo Fassi 175 PDFSebastian AcevedoPas encore d'évaluation