Vous aimerez peut-être aussi
- The Invisible ManDocument22 pagesThe Invisible ManVijay PradhanPas encore d'évaluation
- Atomic SpectraDocument20 pagesAtomic SpectraVijay PradhanPas encore d'évaluation
- Foot Cream Project: Name: - Namrata Pradhan (1215) SypgdpcmDocument8 pagesFoot Cream Project: Name: - Namrata Pradhan (1215) SypgdpcmVijay PradhanPas encore d'évaluation
- Electrochemical SynthesisDocument5 pagesElectrochemical SynthesisVijay PradhanPas encore d'évaluation
- Product Quality Is Directly Proportional To Analytical QualityDocument7 pagesProduct Quality Is Directly Proportional To Analytical QualityVijay PradhanPas encore d'évaluation
- Physicalchemisty: Topic: Nanoarchitecture of Silica and PolydimethysiloxaneDocument27 pagesPhysicalchemisty: Topic: Nanoarchitecture of Silica and PolydimethysiloxaneVijay PradhanPas encore d'évaluation
- Green ChemistryDocument23 pagesGreen ChemistryVijay PradhanPas encore d'évaluation
- Hyphemated TechniquesDocument14 pagesHyphemated TechniquesVijay PradhanPas encore d'évaluation
- Analysis of UrineDocument30 pagesAnalysis of UrineVijay PradhanPas encore d'évaluation
- Auger NewDocument13 pagesAuger NewVijay PradhanPas encore d'évaluation
- Noise Pollution Problem AND SolutionsDocument19 pagesNoise Pollution Problem AND SolutionsVijay PradhanPas encore d'évaluation
- SUB: Physical Chemistry Topic: Solubilization, Micellar Catalysis, Micro Emulsions, Reverse Micelles, Characterization of Micro EmulsionsDocument30 pagesSUB: Physical Chemistry Topic: Solubilization, Micellar Catalysis, Micro Emulsions, Reverse Micelles, Characterization of Micro EmulsionsVijay PradhanPas encore d'évaluation
- T: Q I A C: Opic Uality N Nalytical HemistryDocument13 pagesT: Q I A C: Opic Uality N Nalytical HemistryVijay PradhanPas encore d'évaluation
- Paper 2Document5 pagesPaper 2Vijay PradhanPas encore d'évaluation
- Analytical ChemistryDocument16 pagesAnalytical ChemistryVijay PradhanPas encore d'évaluation
- Analytical ChemistryDocument14 pagesAnalytical ChemistryVijay PradhanPas encore d'évaluation
- Analytical StudyDocument9 pagesAnalytical StudyVijay PradhanPas encore d'évaluation
- Analytical Chemistry.Document18 pagesAnalytical Chemistry.Vijay PradhanPas encore d'évaluation
- Scanning Electron MicrosDocument11 pagesScanning Electron MicrosVijay PradhanPas encore d'évaluation
- Analytical ChemistryDocument16 pagesAnalytical ChemistryVijay PradhanPas encore d'évaluation
- HeterocyclicDocument27 pagesHeterocyclicVijay PradhanPas encore d'évaluation
- Analytical ChemistryDocument14 pagesAnalytical ChemistryVijay PradhanPas encore d'évaluation
- Analytical ChemistryDocument15 pagesAnalytical ChemistryVijay PradhanPas encore d'évaluation
- Polymerization KineticsDocument7 pagesPolymerization KineticsVijay PradhanPas encore d'évaluation
- Basics of IrDocument12 pagesBasics of IrVijay PradhanPas encore d'évaluation
- PhotochemistryDocument24 pagesPhotochemistryVijay PradhanPas encore d'évaluation
- Biomolecules - IIDocument15 pagesBiomolecules - IIVijay PradhanPas encore d'évaluation
- Enamines and YlidesDocument18 pagesEnamines and YlidesVijay Pradhan100% (1)
- Atomic StructureDocument5 pagesAtomic StructureVijay PradhanPas encore d'évaluation
- SymmetryDocument7 pagesSymmetryVijay PradhanPas encore d'évaluation
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- SLA707xM Series: 2-Phase Unipolar Stepper Motor DriverDocument20 pagesSLA707xM Series: 2-Phase Unipolar Stepper Motor DriverKatherine EsperillaPas encore d'évaluation
- 1 Diet Guide PDFDocument43 pages1 Diet Guide PDFYG1Pas encore d'évaluation
- Pathfinder House RulesDocument2 pagesPathfinder House RulesilililiilililliliI100% (1)
- Ass2 mkt1009Document11 pagesAss2 mkt1009thang5423Pas encore d'évaluation
- A Review of The Accounting CycleDocument46 pagesA Review of The Accounting CycleRPas encore d'évaluation
- Đo Nhiệt Độ LI-24ALW-SelectDocument4 pagesĐo Nhiệt Độ LI-24ALW-SelectThang NguyenPas encore d'évaluation
- Q1 LAS 4 FABM2 12 Week 2 3Document7 pagesQ1 LAS 4 FABM2 12 Week 2 3Flare ColterizoPas encore d'évaluation
- Taguig City University: College of Information and Communication TechnologyDocument9 pagesTaguig City University: College of Information and Communication TechnologyRay SenpaiPas encore d'évaluation
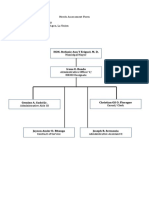
- Needs Assessment Form Company Name: HRMO Address: Sta. Barbara Agoo, La UnionDocument2 pagesNeeds Assessment Form Company Name: HRMO Address: Sta. Barbara Agoo, La UnionAlvin LaroyaPas encore d'évaluation
- Web Development Company in BhubaneswarDocument4 pagesWeb Development Company in BhubaneswarsatyajitPas encore d'évaluation
- Degree 21-22 INTERNSHIPDocument4 pagesDegree 21-22 INTERNSHIPkoushik royalPas encore d'évaluation
- Performance Online - Classic Car Parts CatalogDocument168 pagesPerformance Online - Classic Car Parts CatalogPerformance OnlinePas encore d'évaluation
- BBBB - View ReservationDocument2 pagesBBBB - View ReservationBashir Ahmad BashirPas encore d'évaluation
- Research and Practice in HRM - Sept 8Document9 pagesResearch and Practice in HRM - Sept 8drankitamayekarPas encore d'évaluation
- Makalah MGR Promot - Clma Gravity - p2pd TmminDocument27 pagesMakalah MGR Promot - Clma Gravity - p2pd TmminSandi SaputraPas encore d'évaluation
- Kathrein 739624Document2 pagesKathrein 739624anna.bPas encore d'évaluation
- US Navy Course NAVEDTRA 14342 - Air Traffic ControllerDocument594 pagesUS Navy Course NAVEDTRA 14342 - Air Traffic ControllerGeorges100% (4)
- DSP QBDocument8 pagesDSP QBNithya VijayaPas encore d'évaluation
- Designing A 3D Jewelry ModelDocument4 pagesDesigning A 3D Jewelry ModelAbdulrahman JradiPas encore d'évaluation
- Photoshop TheoryDocument10 pagesPhotoshop TheoryShri BhagwanPas encore d'évaluation
- Bearing SettlementDocument4 pagesBearing SettlementBahaismail100% (1)
- Roger Rabbit:, Forest Town, CA 90020Document3 pagesRoger Rabbit:, Forest Town, CA 90020Marc TPas encore d'évaluation
- Share Purchase Agreement Short FormDocument7 pagesShare Purchase Agreement Short FormGerald HansPas encore d'évaluation
- Lolcat - Linux Cat Command Make Rainbows & Unicorns - LinuxsecretsDocument1 pageLolcat - Linux Cat Command Make Rainbows & Unicorns - LinuxsecretsAli BadPas encore d'évaluation
- SKF BeyondZero White Paper 12761ENDocument12 pagesSKF BeyondZero White Paper 12761ENdiosmio111Pas encore d'évaluation
- Introduction To Risk Management and Insurance 10th Edition Dorfman Test BankDocument26 pagesIntroduction To Risk Management and Insurance 10th Edition Dorfman Test BankMichelleBellsgkb100% (50)
- How To Post Bail For Your Temporary Liberty?Document5 pagesHow To Post Bail For Your Temporary Liberty?Ruel Benjamin Bernaldez100% (1)
- Tutorial Chapter 3Document9 pagesTutorial Chapter 3Sirhan HelmiPas encore d'évaluation
- Hach Company v. In-SituDocument8 pagesHach Company v. In-SituPatent LitigationPas encore d'évaluation
- Encryption LessonDocument2 pagesEncryption LessonKelly LougheedPas encore d'évaluation