Vous aimerez peut-être aussi
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (894)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- 07 01 2024 JR Super60 NUCLEUS BT Jee Adv2021 P1 CTA 23 Q PaperDocument20 pages07 01 2024 JR Super60 NUCLEUS BT Jee Adv2021 P1 CTA 23 Q Paperzaid khanPas encore d'évaluation
- B Pharmacy 2015Document114 pagesB Pharmacy 2015GalataPas encore d'évaluation
- Ammonia and Urea Plants PDFDocument48 pagesAmmonia and Urea Plants PDFbenon100% (1)
- Redox Titration Calculations and AnalysesDocument7 pagesRedox Titration Calculations and AnalysesAtikaRahayuPas encore d'évaluation
- Differential Drive KinematicsDocument8 pagesDifferential Drive KinematicsPratik PatelPas encore d'évaluation
- Enzymes .Document6 pagesEnzymes .Nathan SsekamattePas encore d'évaluation
- Chemistry of The Elements (2nd Edition)Document14 pagesChemistry of The Elements (2nd Edition)mycomiccityPas encore d'évaluation
- Schedule of Rate For Standard Stock Materials Common SR 2021-22 (11 KV System)Document161 pagesSchedule of Rate For Standard Stock Materials Common SR 2021-22 (11 KV System)sagar mukulPas encore d'évaluation
- Heat Transfer PDFDocument3 pagesHeat Transfer PDFTahmeed AzizPas encore d'évaluation
- Aim Is To Investigate Foaming Capacity of Different Washing Soap and Effect of Addition of Sodium Carbonate On ThemDocument3 pagesAim Is To Investigate Foaming Capacity of Different Washing Soap and Effect of Addition of Sodium Carbonate On ThemSachin SoniPas encore d'évaluation
- Page 1 of 3: What To Expect When Being Asked Boiling Point Questions On ExamsDocument3 pagesPage 1 of 3: What To Expect When Being Asked Boiling Point Questions On ExamsSulochana KoviPas encore d'évaluation
- Chapter 7 - Relative Masses and Mole CalculationsDocument9 pagesChapter 7 - Relative Masses and Mole CalculationsAdrawa NorwelPas encore d'évaluation
- BIOL1177 SM1 2020 Session 2 ProformaDocument7 pagesBIOL1177 SM1 2020 Session 2 ProformaThisariePas encore d'évaluation
- Deet1-2 - Lab2 GRP3Document6 pagesDeet1-2 - Lab2 GRP3Jhay lambert MercadoPas encore d'évaluation
- 06 PenetrexDocument13 pages06 PenetrexMari WellPas encore d'évaluation
- Resins For Battery ManufactureDocument4 pagesResins For Battery ManufacturecmscostaPas encore d'évaluation
- PHY 201 - Thermodynamics and Kinetic Theory of Gases: Unit Name of The Unit SyllabusDocument1 pagePHY 201 - Thermodynamics and Kinetic Theory of Gases: Unit Name of The Unit SyllabusBhoomi ShettyPas encore d'évaluation
- Wave Optics Solutions – Fringe Patterns and IntensitiesDocument15 pagesWave Optics Solutions – Fringe Patterns and Intensitiesashu mishraPas encore d'évaluation
- Astm D 6583 00 - Ensaio Padronizado Porosidade Camada de TintaDocument2 pagesAstm D 6583 00 - Ensaio Padronizado Porosidade Camada de TintaNara CamargoPas encore d'évaluation
- EstañoDocument646 pagesEstañoJuan MartínezPas encore d'évaluation
- PH Ysicsguide: Basic Concepts of Statistical MechanicsDocument14 pagesPH Ysicsguide: Basic Concepts of Statistical MechanicsMPas encore d'évaluation
- Industria1 C o M B U S T I o N Pollution and C o N T R o L PDFDocument890 pagesIndustria1 C o M B U S T I o N Pollution and C o N T R o L PDFAfzaal AshrafPas encore d'évaluation
- Jan 23 WCH12 SolvedDocument28 pagesJan 23 WCH12 Solvedthe dsPas encore d'évaluation
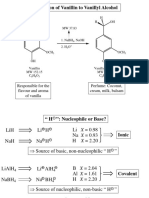
- Síntesis de Alcohol VainillílicoDocument6 pagesSíntesis de Alcohol VainillílicoYago L100% (1)
- Cables: Compensating and ExtensionDocument32 pagesCables: Compensating and ExtensionAyadi_AymanPas encore d'évaluation
- Realization Phase of Model in Terms of EcodesignDocument7 pagesRealization Phase of Model in Terms of EcodesignZvonimir MajićPas encore d'évaluation
- Earth Materials and ProcessesDocument60 pagesEarth Materials and ProcessesZarlene SierraPas encore d'évaluation
- Module 9. Alcohols: Nomenclature, Preparation and ReactionsDocument3 pagesModule 9. Alcohols: Nomenclature, Preparation and ReactionsPeña, RodolfoPas encore d'évaluation
- Cekaman AirDocument4 pagesCekaman AirSaifur RidhalPas encore d'évaluation
- Annealing Process Explained in 40 CharactersDocument8 pagesAnnealing Process Explained in 40 CharactersRohit SharmaPas encore d'évaluation