Vous aimerez peut-être aussi
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (119)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (265)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (399)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (587)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2219)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (894)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (73)
- 2023 Summit Program Draft 5 Apr18Document43 pages2023 Summit Program Draft 5 Apr18Raheem KassamPas encore d'évaluation
- USMLE Step 1 NBME Top Concepts 2021Document475 pagesUSMLE Step 1 NBME Top Concepts 2021dalia khamoPas encore d'évaluation
- Nutrition During InfancyDocument4 pagesNutrition During InfancyMaryHope100% (1)
- Congenital SyphilisDocument28 pagesCongenital SyphilisMeena Koushal100% (4)
- MCQ in MedicineDocument18 pagesMCQ in MedicineEslamAlmassri75% (4)
- Microbiology and Parasitology ReviewerDocument4 pagesMicrobiology and Parasitology ReviewerChrister Jon AcostaPas encore d'évaluation
- Nephrotic Syndrome - NotesDocument22 pagesNephrotic Syndrome - NotesHampson Malekano100% (1)
- Barangay Anti-Smoking OrdinanceDocument4 pagesBarangay Anti-Smoking Ordinancejacquelyn samson100% (1)
- FundaDocument5 pagesFundaGreggy Francisco LaraPas encore d'évaluation
- Feasibility of Muntingia Calabura as WineDocument19 pagesFeasibility of Muntingia Calabura as WineKhen Raselle BaculioPas encore d'évaluation
- Drug Doses 2017Document127 pagesDrug Doses 2017Yuliawati HarunaPas encore d'évaluation
- Link Zoom KONIKA 18-19 OctDocument6 pagesLink Zoom KONIKA 18-19 OctAgus WijataPas encore d'évaluation
- 7869 40826 3 PB PDFDocument8 pages7869 40826 3 PB PDFHendra AjaPas encore d'évaluation
- Pediatric Infectious Diseases Conference Focuses on COVID ManagementDocument2 pagesPediatric Infectious Diseases Conference Focuses on COVID ManagementAgus WijataPas encore d'évaluation
- Pone 0026881Document5 pagesPone 0026881Agus WijataPas encore d'évaluation
- Nna en November09Document3 pagesNna en November09Agus WijataPas encore d'évaluation
- Pediatric Infectious Diseases Conference Focuses on COVID ManagementDocument2 pagesPediatric Infectious Diseases Conference Focuses on COVID ManagementAgus WijataPas encore d'évaluation
- Faktor-Faktor Yang Berhubungan Dengan Kejadian Berat Badan Lahir Rendah (BBLR) Di Kabupaten KudusDocument10 pagesFaktor-Faktor Yang Berhubungan Dengan Kejadian Berat Badan Lahir Rendah (BBLR) Di Kabupaten KudusDellaPas encore d'évaluation
- 7869 40826 3 PB PDFDocument8 pages7869 40826 3 PB PDFHendra AjaPas encore d'évaluation
- Pediatric Infectious Diseases Conference Focuses on COVID ManagementDocument2 pagesPediatric Infectious Diseases Conference Focuses on COVID ManagementAgus WijataPas encore d'évaluation
- Wed SongDocument1 pageWed SongAgus WijataPas encore d'évaluation
- Kids Games Powerpoint TemplateDocument26 pagesKids Games Powerpoint TemplateAgus WijataPas encore d'évaluation
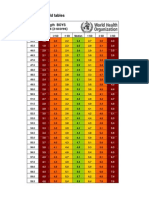
- Boys weight-length z-scores birth to 2 yearsDocument5 pagesBoys weight-length z-scores birth to 2 yearsAgus WijataPas encore d'évaluation
- Mile-Stones of Develovment of ChildrenDocument9 pagesMile-Stones of Develovment of ChildrenAgus WijataPas encore d'évaluation
- Early Biochemical Indicators of Hypoxic-Ischemic Encephalopathy After Birth AsphyxiaDocument5 pagesEarly Biochemical Indicators of Hypoxic-Ischemic Encephalopathy After Birth AsphyxiaAgus WijataPas encore d'évaluation
- Espr Abstracts: Background: Hypoxic-Ischemic Brain Injury (HIE) Is The Most Common Perinatal Cerebral Insult AssociatedDocument1 pageEspr Abstracts: Background: Hypoxic-Ischemic Brain Injury (HIE) Is The Most Common Perinatal Cerebral Insult AssociatedAgus WijataPas encore d'évaluation
- Biochemical Marker As Predictor of Outcome in Perinatal AsphyxiaDocument4 pagesBiochemical Marker As Predictor of Outcome in Perinatal AsphyxiaAgus WijataPas encore d'évaluation
- Status of Serum Bilirubin, Serum Proteins and Prothrombin Time in Babies With Perinatal AsphyxiaDocument4 pagesStatus of Serum Bilirubin, Serum Proteins and Prothrombin Time in Babies With Perinatal AsphyxiaAgus WijataPas encore d'évaluation
- 0 C 96053 C 581 B 042832000000Document1 page0 C 96053 C 581 B 042832000000Agus WijataPas encore d'évaluation
- Edwin Kim, MD A. Wesley Burks, MD Michael Pistiner, MD, MMSCDocument1 pageEdwin Kim, MD A. Wesley Burks, MD Michael Pistiner, MD, MMSCAgus WijataPas encore d'évaluation
- 5211Document7 pages5211Agus WijataPas encore d'évaluation
- CC 100Document4 pagesCC 100Agus WijataPas encore d'évaluation
- Neuron-Specific Enolase As A Marker of The Severity and Outcome of Hypoxic Ischemic EncephalopathyDocument5 pagesNeuron-Specific Enolase As A Marker of The Severity and Outcome of Hypoxic Ischemic EncephalopathyAgus WijataPas encore d'évaluation
- a732b273a10a012b40c8154c371e732bDocument1 pagea732b273a10a012b40c8154c371e732bAgus WijataPas encore d'évaluation
- SDQ Score Sheet and Instructions For Self ReoprtDocument2 pagesSDQ Score Sheet and Instructions For Self ReoprtAgus WijataPas encore d'évaluation
- 0 C 96053 C 581 B 042832000000Document1 page0 C 96053 C 581 B 042832000000Agus WijataPas encore d'évaluation
- Test Definition: NSE: Reporting Title: Neuron Specific Enolase, SDocument1 pageTest Definition: NSE: Reporting Title: Neuron Specific Enolase, SAgus WijataPas encore d'évaluation
- Antimicrob. Agents Chemother. 2015 Ramachandran 1162 7Document6 pagesAntimicrob. Agents Chemother. 2015 Ramachandran 1162 7Agus WijataPas encore d'évaluation
- SDQ Score Sheet and Instructions For Self ReoprtDocument2 pagesSDQ Score Sheet and Instructions For Self ReoprtAgus WijataPas encore d'évaluation
- Apnea NewbornDocument2 pagesApnea NewbornAgus WijataPas encore d'évaluation
- A 55-Year-Old Woman With Shock and Labile Blood PressureDocument11 pagesA 55-Year-Old Woman With Shock and Labile Blood PressureMr. LPas encore d'évaluation
- Cannabis Use and Disorder - Epidemiology, Comorbidity, Health Consequences, and Medico-Legal Status - UpToDateDocument34 pagesCannabis Use and Disorder - Epidemiology, Comorbidity, Health Consequences, and Medico-Legal Status - UpToDateAnonymous kvI7zBNPas encore d'évaluation
- Standards of Care in Child Care InstitutionsDocument36 pagesStandards of Care in Child Care InstitutionsVaishnavi JayakumarPas encore d'évaluation
- AABB Billing Guide For Blood Products and Related Services: July 2020 1Document45 pagesAABB Billing Guide For Blood Products and Related Services: July 2020 1Rija KhanPas encore d'évaluation
- Acute and Chronic PyelonephritisDocument7 pagesAcute and Chronic PyelonephritisMatthew Ryan100% (1)
- The Politics of The Asia-Pacific Triumphs, Challenges, and Threats (Mark S. Williams (Editor)Document381 pagesThe Politics of The Asia-Pacific Triumphs, Challenges, and Threats (Mark S. Williams (Editor)lelenaPas encore d'évaluation
- CHAPTER II Open BurningDocument6 pagesCHAPTER II Open Burningjedric_14100% (1)
- Interstitial Cystitis (Painful Bladder Syndrome) - Causes & TreatmentDocument12 pagesInterstitial Cystitis (Painful Bladder Syndrome) - Causes & TreatmentJimmy GillPas encore d'évaluation
- Expressed Emotion and RelapseDocument31 pagesExpressed Emotion and RelapseshivangifbscPas encore d'évaluation
- Physical Paper +1ANNUALDocument5 pagesPhysical Paper +1ANNUALprabhnoorprimePas encore d'évaluation
- CNH Construction Health DeclarationDocument1 pageCNH Construction Health DeclarationEna Ahmad PiePas encore d'évaluation
- Anti-Rabies Act of 2007 (Ra 9482) 2Document29 pagesAnti-Rabies Act of 2007 (Ra 9482) 2Ronz Rogan100% (1)
- Qigong MassageDocument3 pagesQigong MassageChoi Pei YeePas encore d'évaluation
- Emropub 2016 en 19266Document45 pagesEmropub 2016 en 19266jamshaidjiPas encore d'évaluation
- Progestin-Only Injectables: Characteristics and Health BenefitsDocument11 pagesProgestin-Only Injectables: Characteristics and Health BenefitsRazaria DailynePas encore d'évaluation
- People Like Us (PLUS) Kolkata, Annual Activity Report 2007-08Document18 pagesPeople Like Us (PLUS) Kolkata, Annual Activity Report 2007-08Agniva LahiriPas encore d'évaluation
- To Remember The Four Causes of Cell InjuryDocument43 pagesTo Remember The Four Causes of Cell Injuryapi-3825096Pas encore d'évaluation
- Kirit P. Mehta School of Law, Mumbai: A Project Submitted ONDocument12 pagesKirit P. Mehta School of Law, Mumbai: A Project Submitted ONNikit BaryaPas encore d'évaluation
- WHATs New in CPCRDocument4 pagesWHATs New in CPCRJessicaHernandezPas encore d'évaluation
- Peritoneal DialysisDocument9 pagesPeritoneal Dialysispinkygurlz1990Pas encore d'évaluation