Vous aimerez peut-être aussi
- Bioavailability and Bioequivalence: By: Kris May Lyn A. RamosDocument77 pagesBioavailability and Bioequivalence: By: Kris May Lyn A. RamosValar Morghulis100% (1)
- EcosanoidsDocument21 pagesEcosanoidsfmduniaPas encore d'évaluation
- Drugs in ReptilesDocument71 pagesDrugs in ReptilesSunil100% (1)
- Classification and Dosage of Antimicrobial Agents in Veterinary MedicineDocument24 pagesClassification and Dosage of Antimicrobial Agents in Veterinary MedicineSunil0% (1)
- Drugs Acting On Digestive System of AnimalsDocument11 pagesDrugs Acting On Digestive System of AnimalsSunil100% (3)
- ZOOTOXINSDocument72 pagesZOOTOXINSSunil83% (6)
- PHCL 412-512 MidtermDocument144 pagesPHCL 412-512 Midtermamnguye1100% (1)
- Biopharmaceutical Classification System and Formulation DevelopmentDocument18 pagesBiopharmaceutical Classification System and Formulation DevelopmentAshish Mittal100% (1)
- Prepared By: Murari Pavan M.Pharm (Pharmaceutics) Gautham College of PharmacyDocument22 pagesPrepared By: Murari Pavan M.Pharm (Pharmaceutics) Gautham College of PharmacyAshutosh LohumiPas encore d'évaluation
- 1important DefinitionsDocument3 pages1important DefinitionsBest VideoPas encore d'évaluation
- Inflammation Chronic Diseases and Cancer - Cell and Molecular Biology Immunology and Clinical Bases PDFDocument442 pagesInflammation Chronic Diseases and Cancer - Cell and Molecular Biology Immunology and Clinical Bases PDFsatriomega100% (1)
- Pharmaco DynamicsDocument7 pagesPharmaco DynamicsDavid NicholasPas encore d'évaluation
- Pharma 1.2 - Pharmacokinetics (Wini Ong) PDFDocument11 pagesPharma 1.2 - Pharmacokinetics (Wini Ong) PDFVon Javier GamateroPas encore d'évaluation
- 1introDocument158 pages1introDea MaharanisPas encore d'évaluation
- Principles of Pharmacology Chapter 1Document37 pagesPrinciples of Pharmacology Chapter 1Muhammad ZakriaPas encore d'évaluation
- PharmacologyDocument57 pagesPharmacologyarun231187Pas encore d'évaluation
- Bioassay of DrugsDocument45 pagesBioassay of DrugssivaPas encore d'évaluation
- Pharmacokinetics and Pharmacodynamics 40Document40 pagesPharmacokinetics and Pharmacodynamics 40Shoaib BiradarPas encore d'évaluation
- Basic Pharmacology Notes (Chris Andersen, ICUPrimaryPrep - Com)Document8 pagesBasic Pharmacology Notes (Chris Andersen, ICUPrimaryPrep - Com)PkernPas encore d'évaluation
- Immunotoxicity Studes 2005d 0022 Gdl0001Document13 pagesImmunotoxicity Studes 2005d 0022 Gdl0001Anton MymrikovPas encore d'évaluation
- ImmunotoxicityDocument6 pagesImmunotoxicitysuba_n23100% (1)
- Protein Synthesis InhibitorsDocument25 pagesProtein Synthesis InhibitorsSawsan Z. JwaiedPas encore d'évaluation
- Cagayan State University College of Medicine and Surgery SY: 2009-2010Document41 pagesCagayan State University College of Medicine and Surgery SY: 2009-2010ahmad usmanPas encore d'évaluation
- Drug Extraction RatioDocument8 pagesDrug Extraction RatioHeru Fajar SyaputraPas encore d'évaluation
- Vancomycin: Group 5 R.M 2Document31 pagesVancomycin: Group 5 R.M 2vi_wiviaPas encore d'évaluation
- Trypanosoma: Sri SundariDocument37 pagesTrypanosoma: Sri SundariVaniaPas encore d'évaluation
- ExcellenceDocument26 pagesExcellenceALISHAPas encore d'évaluation
- ANS ReceptorsDocument14 pagesANS ReceptorsAisha AliPas encore d'évaluation
- MacrolidesDocument27 pagesMacrolidesAuthor Nauman Shad100% (1)
- AtropineDocument14 pagesAtropineKrazygopa BalorPas encore d'évaluation
- Excretion of DrugsDocument17 pagesExcretion of DrugsdahiphalehPas encore d'évaluation
- Pharmacology Practical Manual - Student Copy2Document11 pagesPharmacology Practical Manual - Student Copy2NareshPas encore d'évaluation
- Transes PharmacodynamicsDocument36 pagesTranses PharmacodynamicsGwyneth Koleen Lopez100% (1)
- VPT 311: General and Systemic Veterinary Pharmacology (2+1)Document109 pagesVPT 311: General and Systemic Veterinary Pharmacology (2+1)fkw wmkfmPas encore d'évaluation
- Biochemistry JD 2Document6 pagesBiochemistry JD 2failinPas encore d'évaluation
- Brainware University: (BP501T) Class Notes (Medicinal Chemistry-II Theory)Document6 pagesBrainware University: (BP501T) Class Notes (Medicinal Chemistry-II Theory)Mriganka KarmakarPas encore d'évaluation
- 2-Principles of Antimicrobial Therapy 2 PDFDocument29 pages2-Principles of Antimicrobial Therapy 2 PDFShashidharan MenonPas encore d'évaluation
- Drugs Effects On The Central Nervous SystemDocument6 pagesDrugs Effects On The Central Nervous SystemMuhammad AriefPas encore d'évaluation
- Ligand and Cell Signaling PathwaysDocument117 pagesLigand and Cell Signaling PathwaysShoaib MomenPas encore d'évaluation
- Receptors As Drug Targets PDFDocument66 pagesReceptors As Drug Targets PDFAubreyPas encore d'évaluation
- Allergy: Pharm .D, M. Phil Lecturer (Pharmacognosy)Document35 pagesAllergy: Pharm .D, M. Phil Lecturer (Pharmacognosy)Ahmed Imran0% (1)
- ANTI INFLAMMATORY Screening MethodsDocument7 pagesANTI INFLAMMATORY Screening MethodsBrajesh Thankamony67% (3)
- Rickettsia: Introduction, Characteristics, ClassificationDocument5 pagesRickettsia: Introduction, Characteristics, ClassificationMohammed ShahanewzPas encore d'évaluation
- Oxytocin BioassayDocument17 pagesOxytocin BioassayPhArMaCyGrAdUaTeS100% (4)
- Pharmacology Module PDFDocument23 pagesPharmacology Module PDFmirza_baig_46100% (1)
- LocalanestheticsDocument51 pagesLocalanestheticskingkb4uPas encore d'évaluation
- Laboratory Diagnosis in Infections Produced by Anaerobic BacteriaDocument43 pagesLaboratory Diagnosis in Infections Produced by Anaerobic Bacteriarikirdn27Pas encore d'évaluation
- Kinetics Review 2Document37 pagesKinetics Review 2filenotfoundPas encore d'évaluation
- Autocoid PharmacologyDocument29 pagesAutocoid PharmacologyLyadelou Fortu100% (1)
- Uptake and Distribution of Volatile AnestheticsDocument22 pagesUptake and Distribution of Volatile AnestheticsSuresh Kumar100% (3)
- Lecture 1 PDFDocument75 pagesLecture 1 PDFBasil Elbushra Ahmed DomiPas encore d'évaluation
- Nuclear Receptors 2006Document31 pagesNuclear Receptors 2006lsintaningtyasPas encore d'évaluation
- Drug Absorption and DistributionDocument30 pagesDrug Absorption and DistributionaelmowafyPas encore d'évaluation
- General Pharmacology) 343 (Document28 pagesGeneral Pharmacology) 343 (ALNAKI100% (1)
- Blood Banking AssignmentDocument3 pagesBlood Banking AssignmentMia Dela VegaPas encore d'évaluation
- Determination of PKa Values For APIDocument9 pagesDetermination of PKa Values For APISimona Florina Precup100% (1)
- Metabolic DisordersDocument80 pagesMetabolic DisordersXeniyaFedoryakPas encore d'évaluation
- Microbiology Notes Chapers 1-2Document3 pagesMicrobiology Notes Chapers 1-2dinkinpdPas encore d'évaluation
- Autocoids and Their AntagonistsDocument19 pagesAutocoids and Their AntagonistsHossein Sehati100% (1)
- FarmakogenetikaDocument197 pagesFarmakogenetikaAmilaTravnjakPas encore d'évaluation
- K46 Pharmacology of Anthelminthics, Antiprotozoal, & Antimalaria (Farmakologi)Document78 pagesK46 Pharmacology of Anthelminthics, Antiprotozoal, & Antimalaria (Farmakologi)ayapillaiPas encore d'évaluation
- Veterinary Basics1Document119 pagesVeterinary Basics1Saleem Ahmed Shahwani0% (1)
- Intro PK PD Genomics - PharmaDocument110 pagesIntro PK PD Genomics - PharmaKenneth NuñezPas encore d'évaluation
- Antiviral DrugsDocument24 pagesAntiviral DrugsBurhan Nabi0% (1)
- The Leukotrienes: Chemistry and BiologyD'EverandThe Leukotrienes: Chemistry and BiologyLawrence ChakrinPas encore d'évaluation
- Immunomodulation in Domestic Food Animals: Advances in Veterinary Science and Comparative MedicineD'EverandImmunomodulation in Domestic Food Animals: Advances in Veterinary Science and Comparative MedicineBernald CharleyPas encore d'évaluation
- Carbon Monoxide PoisoningDocument22 pagesCarbon Monoxide PoisoningSunilPas encore d'évaluation
- Endocrine Vety PharmaDocument16 pagesEndocrine Vety PharmaSunilPas encore d'évaluation
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- Household Hazards To PetsDocument16 pagesHousehold Hazards To PetsSunilPas encore d'évaluation
- VPT MCQ ForDocument10 pagesVPT MCQ ForSunilPas encore d'évaluation
- Classification of AmphibiansDocument22 pagesClassification of AmphibiansSunilPas encore d'évaluation
- Toxicological Investigation and Its Significance in Animal Health DiagnosisDocument8 pagesToxicological Investigation and Its Significance in Animal Health DiagnosisSunilPas encore d'évaluation
- Fluorosis Phosphorous Nitrate Toxicity in AnimalsDocument55 pagesFluorosis Phosphorous Nitrate Toxicity in AnimalsSunilPas encore d'évaluation
- VCI MSVE 2008 RegulationsDocument136 pagesVCI MSVE 2008 RegulationsSunil50% (2)
- Mercury Lead Arsenic Cadmium ToxicityDocument171 pagesMercury Lead Arsenic Cadmium ToxicitySunilPas encore d'évaluation
- Nsaids and Other Antinflammatory Agents in Veterinary PracticeDocument44 pagesNsaids and Other Antinflammatory Agents in Veterinary PracticeSunil100% (2)
- Drugs Acting On Haematopoietic System of AnimalsDocument28 pagesDrugs Acting On Haematopoietic System of AnimalsSunil100% (1)
- Drugs Acting On Respiratory System of AnimalsDocument8 pagesDrugs Acting On Respiratory System of AnimalsSunil100% (4)
- Antiseptics and Disinfectants For Veterinary ClinicsDocument3 pagesAntiseptics and Disinfectants For Veterinary ClinicsSunil100% (5)
- Drugs in Behavioural Disorders of PetsDocument5 pagesDrugs in Behavioural Disorders of PetsSunilPas encore d'évaluation
- Drugs Acting On Genitourinary System of AnimalsDocument44 pagesDrugs Acting On Genitourinary System of AnimalsSunilPas encore d'évaluation
- Toxicokinetics DynamicsDocument76 pagesToxicokinetics DynamicsSunil100% (1)
- GENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGDocument49 pagesGENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGSunil0% (1)
- Behavioural Modifying Drugs in PetsDocument33 pagesBehavioural Modifying Drugs in PetsSunilPas encore d'évaluation
- LATHYRISM AND PHOTOSENSiTIZATIONDocument33 pagesLATHYRISM AND PHOTOSENSiTIZATIONSunilPas encore d'évaluation
- Fluorosis Phosphorous ToxicityDocument55 pagesFluorosis Phosphorous ToxicitySunilPas encore d'évaluation
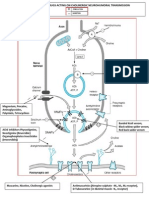
- Site of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFSunilPas encore d'évaluation
- Site of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFSunilPas encore d'évaluation
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- AntiConvulsants Drugs in Brief PDFDocument28 pagesAntiConvulsants Drugs in Brief PDFSunilPas encore d'évaluation
- Environmental PollutantsDocument32 pagesEnvironmental PollutantsSunilPas encore d'évaluation
- Micromeritics 3Document27 pagesMicromeritics 3novi_lingga100% (1)
- Pharmacokinetics: A RefresherDocument27 pagesPharmacokinetics: A RefresherSatriaGafoerPas encore d'évaluation
- Evaluation SeminarDocument29 pagesEvaluation SeminarJolly62Pas encore d'évaluation
- Katzung Chapter 3Document2 pagesKatzung Chapter 3geldevera100% (1)
- Ranitidine Hydrochloride PDFDocument9 pagesRanitidine Hydrochloride PDFDanisha LailaPas encore d'évaluation
- Absorpsi Perkutan Asam SalisilatDocument4 pagesAbsorpsi Perkutan Asam SalisilatINDAHPas encore d'évaluation
- Drug Like PropertiesDocument4 pagesDrug Like PropertiesLe MaPas encore d'évaluation
- Pre FormulationDocument55 pagesPre FormulationEduardo Santos AlquimistaPas encore d'évaluation
- Definitions: - PharmacokineticsDocument31 pagesDefinitions: - PharmacokineticsmebibegPas encore d'évaluation
- New Microsoft Office PowerPoint PresentationDocument27 pagesNew Microsoft Office PowerPoint PresentationGaini ChakravarthyPas encore d'évaluation
- Amorphous Solid Dispersions PDFDocument702 pagesAmorphous Solid Dispersions PDFLuisPas encore d'évaluation
- Pharmacology: By: Nerissa Cabañero Laiza PinedaDocument121 pagesPharmacology: By: Nerissa Cabañero Laiza PinedaJacq CalaycayPas encore d'évaluation
- Gastrointestinal Mucoadhesive Drug Delivery System: A ReviewDocument6 pagesGastrointestinal Mucoadhesive Drug Delivery System: A Reviewcool121086Pas encore d'évaluation
- Preparation, Characterization and In-Vitro Evaluation of Probenecid: A Prototypical Uricosuric Agent in To Extended Release MicrospheresDocument15 pagesPreparation, Characterization and In-Vitro Evaluation of Probenecid: A Prototypical Uricosuric Agent in To Extended Release MicrospheresBaru Chandrasekhar RaoPas encore d'évaluation
- Various Polymer in PharmacyDocument23 pagesVarious Polymer in PharmacyMohamed AbdeshafeePas encore d'évaluation
- Beta-Cyclodextrin Solid DispersionDocument30 pagesBeta-Cyclodextrin Solid Dispersiondarkarva84100% (1)
- Pharmacology Tutorial AnswersDocument2 pagesPharmacology Tutorial AnswerschaiecomelPas encore d'évaluation
- Bioavailability of Diazepam After Intramuscular Injection of Its Water-Soluble Prodrug Alone or With AtropineDocument10 pagesBioavailability of Diazepam After Intramuscular Injection of Its Water-Soluble Prodrug Alone or With AtropineDevinda ArumPas encore d'évaluation
- Review ArticleDocument9 pagesReview ArticlemickydivyaPas encore d'évaluation
- Medicinal Plant PDFDocument37 pagesMedicinal Plant PDFAlka KakkarPas encore d'évaluation
- Biopharmaceutics Classification System-The Scientific Basis PDFDocument5 pagesBiopharmaceutics Classification System-The Scientific Basis PDFtrianawidiacandra100% (1)
- Article 017Document10 pagesArticle 017Digambar GondvalePas encore d'évaluation
- A Renaissance in Peptide TherapeuticsDocument4 pagesA Renaissance in Peptide TherapeuticsReza-ul JalilPas encore d'évaluation
- Solubility, Dissociation or Ionization Constant, Partition Coefficient in PreformulationDocument16 pagesSolubility, Dissociation or Ionization Constant, Partition Coefficient in PreformulationRinta MoonPas encore d'évaluation
- Parental Routes of AdministrationDocument2 pagesParental Routes of AdministrationdrugdrugPas encore d'évaluation
- Biopharmaceutics and Pharmacokinetics in Drug ResearchDocument20 pagesBiopharmaceutics and Pharmacokinetics in Drug Researchlenanazarova1969Pas encore d'évaluation
- Theory of Drug DissolutionDocument96 pagesTheory of Drug DissolutionYuppie RajPas encore d'évaluation
- Pharmacokinetic ParametersDocument29 pagesPharmacokinetic Parametersfaisalnadeem100% (1)