Vous aimerez peut-être aussi
- Clinical ResearchDocument5 pagesClinical ResearchDeepti ShrivasPas encore d'évaluation
- Fast Facts: Clinical Trials in Oncology: The fundamentals of design, conduct and interpretationD'EverandFast Facts: Clinical Trials in Oncology: The fundamentals of design, conduct and interpretationPas encore d'évaluation
- Drug Development: FDA's Definition of A New DrugDocument11 pagesDrug Development: FDA's Definition of A New DrugSophie MendezPas encore d'évaluation
- Principles and Practice of Clinical Trial MedicineD'EverandPrinciples and Practice of Clinical Trial MedicineÉvaluation : 4 sur 5 étoiles4/5 (1)
- Clinical Research NotesDocument98 pagesClinical Research NotesToha Afreen100% (1)
- Pharmacoepidemiology and Pharmacovigilance: Synergistic Tools to Better Investigate Drug SafetyD'EverandPharmacoepidemiology and Pharmacovigilance: Synergistic Tools to Better Investigate Drug SafetyÉvaluation : 4.5 sur 5 étoiles4.5/5 (3)
- The Drug Development ProcessDocument7 pagesThe Drug Development ProcessSACHIN BHASKAR NARKHEDE100% (1)
- Global Clinical Trials Playbook: Capacity and Capability BuildingD'EverandGlobal Clinical Trials Playbook: Capacity and Capability BuildingMenghis BairuPas encore d'évaluation
- Clinical TrialsDocument20 pagesClinical Trialsvamsykrishnabj0% (1)
- The Design and Development of Novel Drugs and Vaccines: Principles and ProtocolsD'EverandThe Design and Development of Novel Drugs and Vaccines: Principles and ProtocolsTarun Kumar BhattPas encore d'évaluation
- NewDocument58 pagesNewAkhil SoodPas encore d'évaluation
- A Textbook of Clinical Research and PharmacovigilanceD'EverandA Textbook of Clinical Research and PharmacovigilanceÉvaluation : 3.5 sur 5 étoiles3.5/5 (3)
- Introduction To Clinical TrialsDocument31 pagesIntroduction To Clinical Trialsmuhammad murtaza88% (8)
- Clinical TrialDocument25 pagesClinical TrialHernawati Bagenda100% (1)
- Clinical Trials in A Nut ShellDocument23 pagesClinical Trials in A Nut ShellSuryamohan SurampudiPas encore d'évaluation
- SEMINAR - 7 (Pharmacoepidemilogy)Document69 pagesSEMINAR - 7 (Pharmacoepidemilogy)LathaVijendraPas encore d'évaluation
- Clinical TrialsDocument39 pagesClinical TrialsYuppie Raj100% (1)
- Drug DevelopmentDocument27 pagesDrug Developmentapi-3810976100% (1)
- Drug Discovery and DevelopmentDocument47 pagesDrug Discovery and DevelopmentSalahBensaber100% (3)
- Preclinical Trials PDFDocument2 pagesPreclinical Trials PDFFiroz TPPas encore d'évaluation
- Clinical Trial DocumentsDocument37 pagesClinical Trial Documentsapi-37446750% (1)
- Clinical Trial DesignsDocument18 pagesClinical Trial DesignsVenkatesh Gavini100% (1)
- Abbreviated New Drug ApplicationDocument32 pagesAbbreviated New Drug Applicationramneet1990100% (1)
- AndaDocument26 pagesAndaNagula Naresh100% (1)
- Clinical Trials PDFDocument17 pagesClinical Trials PDFBin Hip100% (2)
- Schedule YDocument30 pagesSchedule Yapi-3810976100% (13)
- Modern Drug Discovery Process by Sejal KhumanDocument40 pagesModern Drug Discovery Process by Sejal KhumanSejal khuman100% (3)
- Clinical Research OverviewDocument40 pagesClinical Research Overviewjyoti100% (1)
- Investigators BrochureDocument31 pagesInvestigators Brochurepavan_bagga100% (3)
- QA & QC in Clinical TrialDocument54 pagesQA & QC in Clinical Trialpavan_bagga95% (20)
- Pharmacovigilance in Clinical Trials: Version 04 Feb 2021Document81 pagesPharmacovigilance in Clinical Trials: Version 04 Feb 2021Mohammed HammedPas encore d'évaluation
- Vaccine Safety in Pharmacoepidemology and PharmacoeconomicsDocument28 pagesVaccine Safety in Pharmacoepidemology and PharmacoeconomicsGeethika GummadiPas encore d'évaluation
- Guide To Clinical Trial Protocol Content and FormatDocument8 pagesGuide To Clinical Trial Protocol Content and FormatRohit Manakchand ZawarPas encore d'évaluation
- Clinical Trial Essential Documents (Before and During)Document51 pagesClinical Trial Essential Documents (Before and During)Ritika RaginiPas encore d'évaluation
- Ethical Issues in Clinical ResearchDocument23 pagesEthical Issues in Clinical ResearchDaxesh PatelPas encore d'évaluation
- Clinical Research ClassDocument110 pagesClinical Research ClassEreshPas encore d'évaluation
- Protocol Designing in CTDocument5 pagesProtocol Designing in CTSushma Reddy VPas encore d'évaluation
- ICH GCP & Indian Clinical Trial GuidelineDocument97 pagesICH GCP & Indian Clinical Trial GuidelineRanjeet PrasadPas encore d'évaluation
- Phases of Clin TrialDocument26 pagesPhases of Clin TrialPraneeth Sanjeev ReddyPas encore d'évaluation
- Schedule Y GuidelinesDocument49 pagesSchedule Y GuidelinesNaresh Kumar RapoluPas encore d'évaluation
- Study DesignDocument34 pagesStudy DesignVikashgtmPas encore d'évaluation
- Essentail Documents For Conduct of A Clinical TrialDocument26 pagesEssentail Documents For Conduct of A Clinical TrialFarah Aman KhanPas encore d'évaluation
- Clinical Trials Flow ProcessDocument77 pagesClinical Trials Flow ProcessAnonymous Qr9nZRb100% (2)
- Sample Size Estimation in Clinical ResearchDocument9 pagesSample Size Estimation in Clinical ResearchAshish GuptaPas encore d'évaluation
- Drug Safety Associate Job Interview Questions and AnswersDocument8 pagesDrug Safety Associate Job Interview Questions and AnswersGaurav Kumar100% (1)
- Stages of Drug DevelopmentDocument16 pagesStages of Drug DevelopmentVanya MakPas encore d'évaluation
- Ich-Gcp & Schedule yDocument38 pagesIch-Gcp & Schedule ySahiti PendyalaPas encore d'évaluation
- Ind Studies by Sejal KhumanDocument34 pagesInd Studies by Sejal KhumanSejal khumamPas encore d'évaluation
- Types and Designs of Clinical StudiesDocument17 pagesTypes and Designs of Clinical StudiesTrialJoinPas encore d'évaluation
- Importance of Pharmacovigilance For Pharmaceutical IndustryDocument24 pagesImportance of Pharmacovigilance For Pharmaceutical IndustryPiratesPas encore d'évaluation
- 5) PharmacoepidemiologyDocument51 pages5) PharmacoepidemiologyDr. Zirwa AsimPas encore d'évaluation
- Designs of Clinical TrialDocument7 pagesDesigns of Clinical TrialSruthi100% (1)
- Regulatory AffairsDocument14 pagesRegulatory AffairsSiddarth Reddy100% (2)
- E9 - Statistical Principles For Clinical TrialsDocument39 pagesE9 - Statistical Principles For Clinical Trialskappu_abb100% (2)
- PHARMACOVIGILANCEDocument28 pagesPHARMACOVIGILANCENosheen JavedPas encore d'évaluation
- Chapter 2 PEY-Measurement of Outcomes - Pharma DostDocument50 pagesChapter 2 PEY-Measurement of Outcomes - Pharma DostKhadeer AG50% (2)
- Clinical TrialDocument26 pagesClinical TrialFatonyPas encore d'évaluation
- Pharmacovigilance IntroductionDocument29 pagesPharmacovigilance IntroductionMaurizio Sessa100% (1)
- Fluorosis Phosphorous Nitrate Toxicity in AnimalsDocument55 pagesFluorosis Phosphorous Nitrate Toxicity in AnimalsSunilPas encore d'évaluation
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- VPT MCQ ForDocument10 pagesVPT MCQ ForSunilPas encore d'évaluation
- Endocrine Vety PharmaDocument16 pagesEndocrine Vety PharmaSunilPas encore d'évaluation
- Carbon Monoxide PoisoningDocument22 pagesCarbon Monoxide PoisoningSunilPas encore d'évaluation
- Household Hazards To PetsDocument16 pagesHousehold Hazards To PetsSunilPas encore d'évaluation
- VCI MSVE 2008 RegulationsDocument136 pagesVCI MSVE 2008 RegulationsSunil50% (2)
- Classification and Dosage of Antimicrobial Agents in Veterinary MedicineDocument24 pagesClassification and Dosage of Antimicrobial Agents in Veterinary MedicineSunil0% (1)
- Classification of AmphibiansDocument22 pagesClassification of AmphibiansSunilPas encore d'évaluation
- Drugs in Behavioural Disorders of PetsDocument5 pagesDrugs in Behavioural Disorders of PetsSunilPas encore d'évaluation
- Toxicological Investigation and Its Significance in Animal Health DiagnosisDocument8 pagesToxicological Investigation and Its Significance in Animal Health DiagnosisSunilPas encore d'évaluation
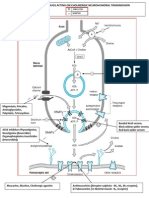
- Site of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Cholinergic Neurohumoral Transmission PDFSunilPas encore d'évaluation
- Drugs in ReptilesDocument71 pagesDrugs in ReptilesSunil100% (1)
- Antiseptics and Disinfectants For Veterinary ClinicsDocument3 pagesAntiseptics and Disinfectants For Veterinary ClinicsSunil100% (5)
- GENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGDocument49 pagesGENERAL LINE of Treatment UREA AMMONIA SALT - poISONINGSunil0% (1)
- Drugs Acting On Haematopoietic System of AnimalsDocument28 pagesDrugs Acting On Haematopoietic System of AnimalsSunil100% (1)
- Nsaids and Other Antinflammatory Agents in Veterinary PracticeDocument44 pagesNsaids and Other Antinflammatory Agents in Veterinary PracticeSunil100% (2)
- Behavioural Modifying Drugs in PetsDocument33 pagesBehavioural Modifying Drugs in PetsSunilPas encore d'évaluation
- Drugs Acting On Digestive System of AnimalsDocument11 pagesDrugs Acting On Digestive System of AnimalsSunil100% (3)
- Drugs Acting On Respiratory System of AnimalsDocument8 pagesDrugs Acting On Respiratory System of AnimalsSunil100% (4)
- Toxicokinetics DynamicsDocument76 pagesToxicokinetics DynamicsSunil100% (1)
- ZOOTOXINSDocument72 pagesZOOTOXINSSunil83% (6)
- LATHYRISM AND PHOTOSENSiTIZATIONDocument33 pagesLATHYRISM AND PHOTOSENSiTIZATIONSunilPas encore d'évaluation
- Drugs Acting On Genitourinary System of AnimalsDocument44 pagesDrugs Acting On Genitourinary System of AnimalsSunilPas encore d'évaluation
- Mercury Lead Arsenic Cadmium ToxicityDocument171 pagesMercury Lead Arsenic Cadmium ToxicitySunilPas encore d'évaluation
- Environmental PollutantsDocument32 pagesEnvironmental PollutantsSunilPas encore d'évaluation
- Fluorosis Phosphorous ToxicityDocument55 pagesFluorosis Phosphorous ToxicitySunilPas encore d'évaluation
- Site of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFDocument1 pageSite of Action of Drugs Acting On Adrenergic Neurohumoral Transmission PDFSunilPas encore d'évaluation
- Mycotoxins in Animal HealthDocument103 pagesMycotoxins in Animal HealthSunil100% (1)
- AntiConvulsants Drugs in Brief PDFDocument28 pagesAntiConvulsants Drugs in Brief PDFSunilPas encore d'évaluation
- Volume1 Eng BioavaiDocument148 pagesVolume1 Eng Bioavaihugoboss_dark100% (1)
- Headache: Migraine and Tension-Type HeadacheDocument12 pagesHeadache: Migraine and Tension-Type HeadacheLoren SangalangPas encore d'évaluation
- CLINDAMYCINDocument1 pageCLINDAMYCINCarlo ToledooPas encore d'évaluation
- Histamine, Serotonin & The Ergot AlkaloidsDocument31 pagesHistamine, Serotonin & The Ergot AlkaloidsnicewanPas encore d'évaluation
- SedativesDocument21 pagesSedativesKeerthana KPas encore d'évaluation
- Test Bank For Pharmacology Clear and Simple A Guide To Drug Classifications and Dosage Calculations 3rd Edition WatkinsDocument6 pagesTest Bank For Pharmacology Clear and Simple A Guide To Drug Classifications and Dosage Calculations 3rd Edition WatkinsJean Taylor100% (39)
- Stereoisomery PharmacologyDocument17 pagesStereoisomery PharmacologyMarcus Vinícius SouzaPas encore d'évaluation
- Indication Expansion Opportunities For Successful Lifecycle ManagementDocument144 pagesIndication Expansion Opportunities For Successful Lifecycle ManagementHerry HendrayadiPas encore d'évaluation
- File 268Document80 pagesFile 268jhansiPas encore d'évaluation
- Cough Syrup Detail:: Prodactive or Expectorant and Suppressant or Dry CoughDocument3 pagesCough Syrup Detail:: Prodactive or Expectorant and Suppressant or Dry CoughFarhadullah KhanPas encore d'évaluation
- (Cholinergic System) Model Questions and AnswersDocument45 pages(Cholinergic System) Model Questions and AnswersAjay SinghPas encore d'évaluation
- Price List All Disc SM - DSM 170622Document1 pagePrice List All Disc SM - DSM 170622Rhesa GuttamaPas encore d'évaluation
- TDM Dan Rancangan Aturan DosisDocument37 pagesTDM Dan Rancangan Aturan Dosisadelin ransunPas encore d'évaluation
- Assessing Pain: Maria Criselda Reicelle Q. Avelino, MAN, RNDocument38 pagesAssessing Pain: Maria Criselda Reicelle Q. Avelino, MAN, RNAbcd TolibasPas encore d'évaluation
- WDR23 Exsum Fin SPDocument70 pagesWDR23 Exsum Fin SPVerónica SilveriPas encore d'évaluation
- OralMeds - ChecklistDocument6 pagesOralMeds - ChecklistXandra BasnilloPas encore d'évaluation
- Internship 1 4Document75 pagesInternship 1 4Mariah Sharmane Juego Santos100% (2)
- Ekatalog 2023 Sulsel RajawaliDocument50 pagesEkatalog 2023 Sulsel RajawaliSafria HamzaPas encore d'évaluation
- Drug Study - Magnesium Sulfate (MGSO4)Document2 pagesDrug Study - Magnesium Sulfate (MGSO4)Leslie LibrandoPas encore d'évaluation
- The Top 15 Best Selling Cancer Drugs in 2022Document16 pagesThe Top 15 Best Selling Cancer Drugs in 2022Soma YasaswiPas encore d'évaluation
- Pediatric Medication Math Review Jan 2011Document7 pagesPediatric Medication Math Review Jan 2011Tyrone Kent HalogPas encore d'évaluation
- Dopamine Antagonist - WikipediaDocument22 pagesDopamine Antagonist - WikipediaMuhammadafif SholehuddinPas encore d'évaluation
- Banned Drugs ListDocument380 pagesBanned Drugs ListKarthik75% (4)
- Atc DDD Tablet Buat Pa IjalDocument57 pagesAtc DDD Tablet Buat Pa IjalAriesta PerwitasariPas encore d'évaluation
- Org MedDocument11 pagesOrg MedTsukishima KeiPas encore d'évaluation
- Recent Advances in Substance Use Disorders.Document29 pagesRecent Advances in Substance Use Disorders.Sachin BaligaPas encore d'évaluation
- 16.MDR-XDR TBDocument18 pages16.MDR-XDR TBLinna SriwaningsiPas encore d'évaluation
- Maximum Recommended Local Anaesthetic Doses For AdultsDocument2 pagesMaximum Recommended Local Anaesthetic Doses For AdultsadithardanaPas encore d'évaluation
- Drugs Study For Surgery WardDocument4 pagesDrugs Study For Surgery WardMariquita BuenafePas encore d'évaluation
- Cialis Tablets in Quetta - Tadalafil Cialis - Cialis 20mgDocument4 pagesCialis Tablets in Quetta - Tadalafil Cialis - Cialis 20mgSana KhanPas encore d'évaluation