Vous aimerez peut-être aussi
- Reading Sub-Test: Part ADocument17 pagesReading Sub-Test: Part AVijayalakshmi Narayanaswami78% (9)
- Antibiotic in OmfsDocument47 pagesAntibiotic in OmfsRajat GuptaPas encore d'évaluation
- Last Minute Revision PointsDocument4 pagesLast Minute Revision PointsShashipriya AgressPas encore d'évaluation
- Drugs Affecting Calcium BalanceDocument63 pagesDrugs Affecting Calcium BalanceRd Chandane100% (1)
- Drug Study TramadolDocument14 pagesDrug Study TramadolBianca Freya Porral85% (13)
- Instant Download Ebook PDF Abnormal Psychology 9th Edition PDF ScribdDocument41 pagesInstant Download Ebook PDF Abnormal Psychology 9th Edition PDF Scribdvictor.lewis791100% (44)
- Al-Hijamah (Cupping) - Healing The Sunnah Way - Amjad Ahsan AliDocument194 pagesAl-Hijamah (Cupping) - Healing The Sunnah Way - Amjad Ahsan Alihal bannaPas encore d'évaluation
- Bleeding DisordersDocument137 pagesBleeding DisordersJosiah BimabamPas encore d'évaluation
- Uworld JournalDocument3 pagesUworld JournalJayPas encore d'évaluation
- Principles of Antibiotics: Deepa V Post Graduate Student DSCDS, BangaloreDocument117 pagesPrinciples of Antibiotics: Deepa V Post Graduate Student DSCDS, BangaloreDeepa VenkateshPas encore d'évaluation
- NEPHROTIC SYNDROME - HamidDocument20 pagesNEPHROTIC SYNDROME - HamidAbdul Hamid OmarPas encore d'évaluation
- Complications of Local Anesthesia: DR Amna Muzaffar BDS, Fcps Assistant Professor, OmfsDocument74 pagesComplications of Local Anesthesia: DR Amna Muzaffar BDS, Fcps Assistant Professor, OmfsAbdul MananPas encore d'évaluation
- FINAL Hypertension Medication Summary SSDocument1 pageFINAL Hypertension Medication Summary SSronique reidPas encore d'évaluation
- Cranial Nerves With Emphasis ON Facial NerveDocument43 pagesCranial Nerves With Emphasis ON Facial Nervepriti adsulPas encore d'évaluation
- A Summary of The Chemical Mediators Involve in The Acute Inflammatory Response Is Shown in The Table BelowDocument30 pagesA Summary of The Chemical Mediators Involve in The Acute Inflammatory Response Is Shown in The Table Belowinny100% (1)
- Critical Care: Version 2 (2020)Document56 pagesCritical Care: Version 2 (2020)yazzPas encore d'évaluation
- Adverse Drug EffectsDocument66 pagesAdverse Drug EffectsSuba Ranjana BalaPas encore d'évaluation
- Medical Emergency in DentistryDocument42 pagesMedical Emergency in Dentistrywaseem quazi100% (3)
- Surgery 2012Document28 pagesSurgery 2012max_21ruPas encore d'évaluation
- Acute Urinary ObstructionDocument34 pagesAcute Urinary ObstructionHafizur RashidPas encore d'évaluation
- Allergic RhinitisDocument20 pagesAllergic RhinitisMuhammed SuffianPas encore d'évaluation
- PharmacologyDocument120 pagesPharmacologyFluffy_icePas encore d'évaluation
- Normal Laboratory Values With Nursing Consideration - UsnganDocument8 pagesNormal Laboratory Values With Nursing Consideration - UsnganPrincess Nasima M. UsnganPas encore d'évaluation
- Differential Diagnosis of Common ComplaintsDocument105 pagesDifferential Diagnosis of Common ComplaintsJeffTaborPas encore d'évaluation
- ENT Quick ReviewDocument6 pagesENT Quick ReviewWade100% (1)
- ShockDocument21 pagesShockMin-Joo Esther ParkPas encore d'évaluation
- Male GU ExamDocument5 pagesMale GU ExamOmar Farid ElgebalyPas encore d'évaluation
- Version 2 (2020) :,, With The Most Recent Recalls and The UK GuidelinesDocument65 pagesVersion 2 (2020) :,, With The Most Recent Recalls and The UK Guidelinesahmad abugararaPas encore d'évaluation
- Pharmacology of The EyeDocument8 pagesPharmacology of The EyeRahul IyerPas encore d'évaluation
- Nephrotic Syndrome - Armando HasudunganDocument18 pagesNephrotic Syndrome - Armando HasudunganzahraaPas encore d'évaluation
- Leg UlcerDocument28 pagesLeg UlcerRanindya PutriPas encore d'évaluation
- HYPOCALCEMIADocument27 pagesHYPOCALCEMIAJeffri SetiawanPas encore d'évaluation
- Cranial Nerves: DR - Ahmed Gaber Ass. Prof of Neurology Ain Shams UniversityDocument45 pagesCranial Nerves: DR - Ahmed Gaber Ass. Prof of Neurology Ain Shams UniversityKhaled OssamaPas encore d'évaluation
- Neurology - Saif.wesmosis.2013 104907Document68 pagesNeurology - Saif.wesmosis.2013 104907Helene AlawamiPas encore d'évaluation
- EndocrineDocument23 pagesEndocrinensvickneswaranPas encore d'évaluation
- Ophthalmology - Passmedicine 2012 - 62013146Document18 pagesOphthalmology - Passmedicine 2012 - 62013146abuahmed&janaPas encore d'évaluation
- Headache NewDocument17 pagesHeadache NewHaris PeacePas encore d'évaluation
- Significance of History Taking Oral SurgeryDocument32 pagesSignificance of History Taking Oral SurgeryFourthMolar.com0% (1)
- Approach To Unconscious PatientDocument23 pagesApproach To Unconscious Patienttantw880% (1)
- WHO Analgesic LadderDocument1 pageWHO Analgesic LadderJhop SantosPas encore d'évaluation
- Psoriasis Dermatitis FinalDocument50 pagesPsoriasis Dermatitis Finalapi-546809761Pas encore d'évaluation
- Check Unit 557 Jan-Feb Genetics v3 PDFDocument36 pagesCheck Unit 557 Jan-Feb Genetics v3 PDFdragon66Pas encore d'évaluation
- Connective Tissue DiseasesDocument54 pagesConnective Tissue DiseasesRatnakar KamathPas encore d'évaluation
- Morphology of Cell Injury WordDocument8 pagesMorphology of Cell Injury WordNCPP 2K18Pas encore d'évaluation
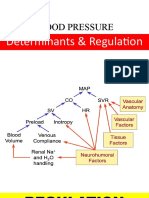
- Short and Long Term Regulation and Determinants of BPDocument90 pagesShort and Long Term Regulation and Determinants of BPDan Tristam MicabaloPas encore d'évaluation
- Principles of Antiplatelet Therapy: DR Htet Htet Htethtet@Imu - Edu.MyDocument36 pagesPrinciples of Antiplatelet Therapy: DR Htet Htet Htethtet@Imu - Edu.MyAbby Liew100% (1)
- Dentistry EmergenciesDocument5 pagesDentistry EmergenciesSofia PereiraPas encore d'évaluation
- Anatomy of Thyroid GlandDocument12 pagesAnatomy of Thyroid GlandBinbinbabu BinuPas encore d'évaluation
- Cholinergic Anticholinergic DrugsDocument60 pagesCholinergic Anticholinergic DrugsMD. RASEL MAHMUD MIMPas encore d'évaluation
- Kuliah SuturingDocument69 pagesKuliah SuturingiqiqiqiqiqPas encore d'évaluation
- Local Anesthesia I LectureDocument44 pagesLocal Anesthesia I LectureMavisPas encore d'évaluation
- C - VVV VV VVVV VVV - VVV VV - VVVV VV VVDocument3 pagesC - VVV VV VVVV VVV - VVV VV - VVVV VV VVBea Angela Bithao AnonoyPas encore d'évaluation
- Coagulation Disorders in ObsDocument33 pagesCoagulation Disorders in Obsapule geraldhumblePas encore d'évaluation
- BMJ - Hypovolemic ShockDocument5 pagesBMJ - Hypovolemic ShockSamer Darwiche Yasiin100% (1)
- Pleural DiseasesDocument4 pagesPleural DiseasesJennifer DayPas encore d'évaluation
- Facial NerveDocument65 pagesFacial NerveprashanthsumsaiarjunPas encore d'évaluation
- Diagnoses and Management Acute Headache Emergency DepartmentDocument37 pagesDiagnoses and Management Acute Headache Emergency DepartmentBendy Dwi IrawanPas encore d'évaluation
- Peri-Operative Management of Patients Receiving AnticoagulantsDocument22 pagesPeri-Operative Management of Patients Receiving AnticoagulantsCheuk Hei LauPas encore d'évaluation
- MUT17 Booklet 2Document18 pagesMUT17 Booklet 2baang43farhanPas encore d'évaluation
- Venous ThromboembolismDocument49 pagesVenous Thromboembolismadamu mohammadPas encore d'évaluation
- Lowe Syndrome (Oculocerebrorenal syndrome) A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsD'EverandLowe Syndrome (Oculocerebrorenal syndrome) A Simple Guide To The Condition, Diagnosis, Treatment And Related ConditionsPas encore d'évaluation
- Manual of First Aid Professional English: Part 3—Case StudiesD'EverandManual of First Aid Professional English: Part 3—Case StudiesPas encore d'évaluation
- Gastrointestinal Examination - DR - Hammouri's Style: Rahaf Wardeh Internal Medicine 2016-2017Document5 pagesGastrointestinal Examination - DR - Hammouri's Style: Rahaf Wardeh Internal Medicine 2016-2017asdddPas encore d'évaluation
- Louis PasteurDocument27 pagesLouis Pasteurjovmac2uPas encore d'évaluation
- Understanding Tick Bites and Lyme DiseaseDocument2 pagesUnderstanding Tick Bites and Lyme DiseasejeffPas encore d'évaluation
- Other Economically Important Species of Piper: Indian Institute of Spices Research, Kozhikode-673012, Kerala, IndiaDocument13 pagesOther Economically Important Species of Piper: Indian Institute of Spices Research, Kozhikode-673012, Kerala, IndiaAleksandra MilenkovićPas encore d'évaluation
- Advance DirectivesDocument4 pagesAdvance Directiveshaxa yzaPas encore d'évaluation
- Chest X Ray BasicsDocument99 pagesChest X Ray BasicsHarshaWakodkarPas encore d'évaluation
- Preventive Veterinary Medicine PDFDocument48 pagesPreventive Veterinary Medicine PDFMasum Rana100% (2)
- Obesity & Anaesthesia: Co-Ordinator - Dr. Chavi Sethi (MD) Speaker - Dr. Uday Pratap SinghDocument56 pagesObesity & Anaesthesia: Co-Ordinator - Dr. Chavi Sethi (MD) Speaker - Dr. Uday Pratap SinghRafi ramdhanPas encore d'évaluation
- CH11 Patho Benitado 405 - 407Document17 pagesCH11 Patho Benitado 405 - 407Joy VelascoPas encore d'évaluation
- Manju Mehta, Rajesh Sagar (Eds.) - A Practical Approach To Cognitive Behaviour Therapy For Adolescents-Springer India (2015) PDFDocument429 pagesManju Mehta, Rajesh Sagar (Eds.) - A Practical Approach To Cognitive Behaviour Therapy For Adolescents-Springer India (2015) PDFdardoPas encore d'évaluation
- General Declaration Outward/Inward: Departure PlaceDocument1 pageGeneral Declaration Outward/Inward: Departure Placebetty anPas encore d'évaluation
- Review QuestionsDocument266 pagesReview QuestionsBenjoe Ilagan50% (4)
- Antibiotic Treatment For Newborns With Congenital Syphilis (Review)Document47 pagesAntibiotic Treatment For Newborns With Congenital Syphilis (Review)link_wolfloboPas encore d'évaluation
- May Sumotuwethfrsa May Is The Fifth Month of The Year in The Julian and Gregorian Calendars and TheDocument18 pagesMay Sumotuwethfrsa May Is The Fifth Month of The Year in The Julian and Gregorian Calendars and TheRadovan SpiridonovPas encore d'évaluation
- Jeronimus PersonalityandtheCoronavirusPandemicDocument107 pagesJeronimus PersonalityandtheCoronavirusPandemicEmmaCorovicPas encore d'évaluation
- DocxDocument53 pagesDocxkuro hanabusaPas encore d'évaluation
- Stroke Associated With COVID-19 VaccinesDocument23 pagesStroke Associated With COVID-19 VaccinesTUTO TUTOPas encore d'évaluation
- Management of Acute Abdomen in Pregnancy Current PerspectivesDocument16 pagesManagement of Acute Abdomen in Pregnancy Current PerspectivesjessicapxePas encore d'évaluation
- Lepy 103Document19 pagesLepy 103Syed FerozPas encore d'évaluation
- Latihan EkspertiseDocument40 pagesLatihan EkspertiseRoberto HutapeaPas encore d'évaluation
- (Non Trauma) 24-09-2023 - TN - Kalil - DOC Ec CVA ICHDocument11 pages(Non Trauma) 24-09-2023 - TN - Kalil - DOC Ec CVA ICHRasyidu MashuriPas encore d'évaluation
- Ehlers-Danlos Panel: ExpertDocument17 pagesEhlers-Danlos Panel: ExpertMalika ButtPas encore d'évaluation
- Abnormal Psychology VocabularyDocument8 pagesAbnormal Psychology Vocabularym i l k t e a .Pas encore d'évaluation
- DD of Acute Kidney Injury: Mobashirul Islam - Roll No. 91 Moksha Manglani - Roll No. 92Document17 pagesDD of Acute Kidney Injury: Mobashirul Islam - Roll No. 91 Moksha Manglani - Roll No. 92122ritik goyalPas encore d'évaluation
- Picrorhiza KurroaDocument13 pagesPicrorhiza KurroaPiks DhPas encore d'évaluation
- Latihan Soal Soal News ItemDocument2 pagesLatihan Soal Soal News ItemKasmawati YonnediPas encore d'évaluation