Vous aimerez peut-être aussi
- Hidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceD'EverandHidden Figures: The American Dream and the Untold Story of the Black Women Mathematicians Who Helped Win the Space RaceÉvaluation : 4 sur 5 étoiles4/5 (895)
- Never Split the Difference: Negotiating As If Your Life Depended On ItD'EverandNever Split the Difference: Negotiating As If Your Life Depended On ItÉvaluation : 4.5 sur 5 étoiles4.5/5 (838)
- Assistant Prof. D. Eman Khammas AI-sadi Pediatrician /community Medicine DepartmentDocument20 pagesAssistant Prof. D. Eman Khammas AI-sadi Pediatrician /community Medicine DepartmentEman KhammasPas encore d'évaluation
- Nutrition: Assistant Prof. D. Eman Khammas AI-sadi Pediatrician /community Medicine DepartmentDocument25 pagesNutrition: Assistant Prof. D. Eman Khammas AI-sadi Pediatrician /community Medicine DepartmentEman KhammasPas encore d'évaluation
- Vitamins L5Document22 pagesVitamins L5Eman KhammasPas encore d'évaluation
- Lecture 1 IntroductionDocument27 pagesLecture 1 IntroductionEman KhammasPas encore d'évaluation
- Protein L4Document24 pagesProtein L4Eman KhammasPas encore d'évaluation
- Cho L2Document23 pagesCho L2Eman KhammasPas encore d'évaluation
- Dr. Eman Khammas Al-Aadi Head of Community Medicine DepartmentDocument19 pagesDr. Eman Khammas Al-Aadi Head of Community Medicine DepartmentEman KhammasPas encore d'évaluation
- Epidemiology Lectures 3,4: Dr. Eman Khammas Al-Sadi Assistant Prof. in Pediatrics Chief of Community Medicine DepartmentDocument20 pagesEpidemiology Lectures 3,4: Dr. Eman Khammas Al-Sadi Assistant Prof. in Pediatrics Chief of Community Medicine DepartmentEman KhammasPas encore d'évaluation
- Analytical Epidemiology: Dr. Eman Khammas Al-Sadi Chief of Community DepartmentDocument23 pagesAnalytical Epidemiology: Dr. Eman Khammas Al-Sadi Chief of Community DepartmentEman KhammasPas encore d'évaluation
- Epidemiological Data: Dr. Eman Khammas Alsadi Head of Community Medicine DepartmentDocument23 pagesEpidemiological Data: Dr. Eman Khammas Alsadi Head of Community Medicine DepartmentEman KhammasPas encore d'évaluation
- Pathology Parathyroid GlandsDocument14 pagesPathology Parathyroid GlandsEman KhammasPas encore d'évaluation
- MumpsDocument8 pagesMumpsEman KhammasPas encore d'évaluation
- Infant of Diabetic Mother: DR: Eman Khammas Al-SadiDocument60 pagesInfant of Diabetic Mother: DR: Eman Khammas Al-SadiEman KhammasPas encore d'évaluation
- 9 Diabetes Mellitus PHYSIOLOGYDocument22 pages9 Diabetes Mellitus PHYSIOLOGYEman KhammasPas encore d'évaluation
- Anaemia Blood QuestionnaireDocument3 pagesAnaemia Blood QuestionnaireEman KhammasPas encore d'évaluation
- Lecture 1 IntroductionDocument27 pagesLecture 1 IntroductionEman KhammasPas encore d'évaluation
- Physical HazardsDocument18 pagesPhysical HazardsEman KhammasPas encore d'évaluation
- Lebnan GuidlinesDocument36 pagesLebnan GuidlinesEman KhammasPas encore d'évaluation
- ChickenpoxDocument7 pagesChickenpoxEman KhammasPas encore d'évaluation
- 25th Lecture 25 Integumentary System-1Document24 pages25th Lecture 25 Integumentary System-1Eman KhammasPas encore d'évaluation
- ChickenpoxDocument7 pagesChickenpoxEman KhammasPas encore d'évaluation
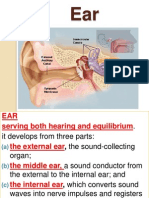
- 23rd EarDocument23 pages23rd EarEman KhammasPas encore d'évaluation
- Student Nam1Document2 pagesStudent Nam1Eman KhammasPas encore d'évaluation
- The Yellow House: A Memoir (2019 National Book Award Winner)D'EverandThe Yellow House: A Memoir (2019 National Book Award Winner)Évaluation : 4 sur 5 étoiles4/5 (98)
- The Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeD'EverandThe Subtle Art of Not Giving a F*ck: A Counterintuitive Approach to Living a Good LifeÉvaluation : 4 sur 5 étoiles4/5 (5794)
- Shoe Dog: A Memoir by the Creator of NikeD'EverandShoe Dog: A Memoir by the Creator of NikeÉvaluation : 4.5 sur 5 étoiles4.5/5 (537)
- Devil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaD'EverandDevil in the Grove: Thurgood Marshall, the Groveland Boys, and the Dawn of a New AmericaÉvaluation : 4.5 sur 5 étoiles4.5/5 (266)
- The Little Book of Hygge: Danish Secrets to Happy LivingD'EverandThe Little Book of Hygge: Danish Secrets to Happy LivingÉvaluation : 3.5 sur 5 étoiles3.5/5 (400)
- Elon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureD'EverandElon Musk: Tesla, SpaceX, and the Quest for a Fantastic FutureÉvaluation : 4.5 sur 5 étoiles4.5/5 (474)
- A Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryD'EverandA Heartbreaking Work Of Staggering Genius: A Memoir Based on a True StoryÉvaluation : 3.5 sur 5 étoiles3.5/5 (231)
- Grit: The Power of Passion and PerseveranceD'EverandGrit: The Power of Passion and PerseveranceÉvaluation : 4 sur 5 étoiles4/5 (588)
- The Emperor of All Maladies: A Biography of CancerD'EverandThe Emperor of All Maladies: A Biography of CancerÉvaluation : 4.5 sur 5 étoiles4.5/5 (271)
- The Unwinding: An Inner History of the New AmericaD'EverandThe Unwinding: An Inner History of the New AmericaÉvaluation : 4 sur 5 étoiles4/5 (45)
- On Fire: The (Burning) Case for a Green New DealD'EverandOn Fire: The (Burning) Case for a Green New DealÉvaluation : 4 sur 5 étoiles4/5 (74)
- The Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersD'EverandThe Hard Thing About Hard Things: Building a Business When There Are No Easy AnswersÉvaluation : 4.5 sur 5 étoiles4.5/5 (344)
- Team of Rivals: The Political Genius of Abraham LincolnD'EverandTeam of Rivals: The Political Genius of Abraham LincolnÉvaluation : 4.5 sur 5 étoiles4.5/5 (234)
- The Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreD'EverandThe Gifts of Imperfection: Let Go of Who You Think You're Supposed to Be and Embrace Who You AreÉvaluation : 4 sur 5 étoiles4/5 (1090)
- The World Is Flat 3.0: A Brief History of the Twenty-first CenturyD'EverandThe World Is Flat 3.0: A Brief History of the Twenty-first CenturyÉvaluation : 3.5 sur 5 étoiles3.5/5 (2259)
- The Sympathizer: A Novel (Pulitzer Prize for Fiction)D'EverandThe Sympathizer: A Novel (Pulitzer Prize for Fiction)Évaluation : 4.5 sur 5 étoiles4.5/5 (121)
- Her Body and Other Parties: StoriesD'EverandHer Body and Other Parties: StoriesÉvaluation : 4 sur 5 étoiles4/5 (821)
- Factors Affecting Mental HealthDocument4 pagesFactors Affecting Mental HealthSamantha CorpuzPas encore d'évaluation
- The Learning Curve For Laparoscopic Cholecystectomy PDFDocument5 pagesThe Learning Curve For Laparoscopic Cholecystectomy PDFIgor CemortanPas encore d'évaluation
- Treatment For Stage IV Cancer PatientsDocument40 pagesTreatment For Stage IV Cancer Patientsk0j1l0100% (1)
- BookBalkanMedicalWeek FINALDocument48 pagesBookBalkanMedicalWeek FINALsome bodyPas encore d'évaluation
- Prittie-2006-Journal of Veterinary Emergency and Critical CareDocument11 pagesPrittie-2006-Journal of Veterinary Emergency and Critical Carejose fdoPas encore d'évaluation
- Stages of LaborDocument51 pagesStages of LaborZeen_Zeen_Fern_3128100% (4)
- Baileys 13th Ed Chapter QuestionsDocument79 pagesBaileys 13th Ed Chapter QuestionsrhymePas encore d'évaluation
- Photob 2022 0067Document10 pagesPhotob 2022 0067Marilene da Conceicao Felix da SilvaPas encore d'évaluation
- Potential of rDNA Technology in Revolutionizing FutureDocument23 pagesPotential of rDNA Technology in Revolutionizing FutureNusra FaizPas encore d'évaluation
- Mechanism of HyperventilationDocument1 pageMechanism of HyperventilationEdio PathicPas encore d'évaluation
- HemodynamicsDocument40 pagesHemodynamicsBravo HeroPas encore d'évaluation
- Spike Protein Detox GuideDocument3 pagesSpike Protein Detox GuideMauro CabelloPas encore d'évaluation
- Look Alike Sound Alike Drug ListDocument1 pageLook Alike Sound Alike Drug ListmohammedPas encore d'évaluation
- Iatrogenic Injury To The Internal Iliac Vein 2008Document4 pagesIatrogenic Injury To The Internal Iliac Vein 2008waldemar russellPas encore d'évaluation
- TabunDocument24 pagesTabunMiftakul SururiPas encore d'évaluation
- KayachikitsaDocument14 pagesKayachikitsasivva1986Pas encore d'évaluation
- Ankylosing SpondylitisDocument5 pagesAnkylosing SpondylitisHarry IsraPas encore d'évaluation
- Laparoscopic AdrenalectomyDocument21 pagesLaparoscopic AdrenalectomyHu EyongPas encore d'évaluation
- Case 1 - Kasey KoxDocument6 pagesCase 1 - Kasey Koxface facePas encore d'évaluation
- MIcro Bio Case StudyDocument3 pagesMIcro Bio Case StudyApex Torres0% (1)
- Self Review.Document7 pagesSelf Review.ulcPas encore d'évaluation
- What Is BPC 157Document2 pagesWhat Is BPC 157MavisPas encore d'évaluation
- Hes 032 Lec Long Quiz 1 BSN 2 A11Document24 pagesHes 032 Lec Long Quiz 1 BSN 2 A11Sabrina MascardoPas encore d'évaluation
- Growth - Development of Fetus - NeonateDocument40 pagesGrowth - Development of Fetus - NeonatesujidahPas encore d'évaluation
- Bio PoemDocument3 pagesBio Poemapi-4039173890% (1)
- Opalescence Boost 40 PercentDocument4 pagesOpalescence Boost 40 PercentAnthony GamarraPas encore d'évaluation
- Microbiota-Targeted Interventions For Mental Health: ReviewDocument7 pagesMicrobiota-Targeted Interventions For Mental Health: Reviewsamira6alvarado6zebaPas encore d'évaluation
- The Problem of Validity in Field Studies of Psychological Disorders Revisited 1990Document14 pagesThe Problem of Validity in Field Studies of Psychological Disorders Revisited 1990FiorellaPas encore d'évaluation
- Leukemia StudyDocument18 pagesLeukemia StudyaneeshdPas encore d'évaluation
- TH TH THDocument5 pagesTH TH THRomelu MartialPas encore d'évaluation