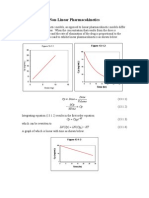
Vous aimerez peut-être aussi
- -Document66 pages-Razor11111Pas encore d'évaluation
- Pka, Log P, Log D and AbsorptionDocument31 pagesPka, Log P, Log D and Absorptiondivyenshah3Pas encore d'évaluation
- U1P4 Experiments On Guinea Pig IleumDocument14 pagesU1P4 Experiments On Guinea Pig IleumMatt StowPas encore d'évaluation
- QSAR and Drug Design: New Developments and ApplicationsD'EverandQSAR and Drug Design: New Developments and ApplicationsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Chapter III Pharmacokinetics: Durge Raj GhalanDocument64 pagesChapter III Pharmacokinetics: Durge Raj GhalanDurge Raj Ghalan100% (3)
- Factors Influencing GI Absorption of DrugDocument11 pagesFactors Influencing GI Absorption of DrugjeorajaPas encore d'évaluation
- Dosage Form Design: Pharmaceutical ConsiderationsDocument9 pagesDosage Form Design: Pharmaceutical ConsiderationsVIJAY KUMAR TIRUKKACHIPas encore d'évaluation
- Drug Elimination KineticsDocument12 pagesDrug Elimination KineticsBenjel AndayaPas encore d'évaluation
- Drug Absorption: - Absorption Is The Process byDocument57 pagesDrug Absorption: - Absorption Is The Process byccccccc1Pas encore d'évaluation
- Pharmacokinetics AbsorptionDocument27 pagesPharmacokinetics AbsorptionchondroboraPas encore d'évaluation
- BASIC PHARMACOKINETICS - CHAPTER 15: Exams IIDocument5 pagesBASIC PHARMACOKINETICS - CHAPTER 15: Exams IIDrHeba100% (1)
- 03 - Bioavailability - Physicochemical and Dosage Form FactorsDocument81 pages03 - Bioavailability - Physicochemical and Dosage Form FactorsBio Data100% (1)
- Drug MetabolismDocument78 pagesDrug Metabolismjuz gonzagaPas encore d'évaluation
- Pharmaceutical Calc ExamDocument15 pagesPharmaceutical Calc ExamMickey Brown100% (2)
- Introduction To Pharmaceutics: (Dosage Forms and Routes of Drug Administration)Document21 pagesIntroduction To Pharmaceutics: (Dosage Forms and Routes of Drug Administration)samiveni50% (2)
- Practical Lab Manual for Pharmaceutical ExperimentsDocument15 pagesPractical Lab Manual for Pharmaceutical ExperimentsMahesh Chougule100% (1)
- 5 Biopharmaceutic Considerations of A Drug DesignDocument89 pages5 Biopharmaceutic Considerations of A Drug DesignImDanaBananaaaPas encore d'évaluation
- Physicochemical Propirtites of AbsorptionDocument32 pagesPhysicochemical Propirtites of AbsorptionBandameedi RamuPas encore d'évaluation
- Steroids: Pharmacognosy #6Document23 pagesSteroids: Pharmacognosy #6Arman Renz PauloPas encore d'évaluation
- BioisosterismDocument22 pagesBioisosterismpurnima singhPas encore d'évaluation
- Spectro ParacetamolDocument5 pagesSpectro ParacetamolAdang FirmansyahPas encore d'évaluation
- A. Principles of Drug MetabolismDocument4 pagesA. Principles of Drug MetabolismLynette EvangelistaPas encore d'évaluation
- Gatwech Dech RutDocument16 pagesGatwech Dech RutGatwech DechPas encore d'évaluation
- 07 Dosage RegimenDocument44 pages07 Dosage Regimenzetttttttttt100% (3)
- Isosterism and BioisosterismDocument7 pagesIsosterism and Bioisosterismmezuniga1100% (1)
- Pharmacotherapy of HTNDocument57 pagesPharmacotherapy of HTNAbera JamboPas encore d'évaluation
- Pharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TDocument21 pagesPharamcokinetics: Course In-Charge: Nimra Waheed Course Name: Biopharmaceutics and Pharmacokinetics Course Code: 613-TNeha GulfamPas encore d'évaluation
- SAR of benzodiazepines structure-activity relationshipsDocument8 pagesSAR of benzodiazepines structure-activity relationshipsSomnath MondalPas encore d'évaluation
- SAR of Benzodizepines by Dr.P.B. MohiteDocument20 pagesSAR of Benzodizepines by Dr.P.B. MohitePRITAM ACHARJYAPas encore d'évaluation
- Key Pharmacokinetic CalculationsDocument10 pagesKey Pharmacokinetic CalculationsMuqaddam Ahmed SalimPas encore d'évaluation
- Medicinal Chemistry 1 - Drug MetabolismDocument39 pagesMedicinal Chemistry 1 - Drug MetabolismPark arimaPas encore d'évaluation
- FlavonoidsDocument23 pagesFlavonoidsunkingkakarotPas encore d'évaluation
- Flavanoids, L 3Document48 pagesFlavanoids, L 3Ammadazfar ImamPas encore d'évaluation
- Medicinal Chemistry of Beta-Lactam AntibioticsDocument13 pagesMedicinal Chemistry of Beta-Lactam AntibioticsJosiah O OmobaPas encore d'évaluation
- BASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDocument22 pagesBASIC PHARMACOKINETICS - CHAPTER 13: Non-Linear KineticsDrHeba100% (7)
- DPCODocument30 pagesDPCOArya SreedharanPas encore d'évaluation
- Qsar 1Document34 pagesQsar 1Srigiriraju VedavyasPas encore d'évaluation
- B.P.P.K - Course OutcomesDocument3 pagesB.P.P.K - Course OutcomesDr. B. Sree Giri PrasadPas encore d'évaluation
- C-9 Modified ReleaseDocument6 pagesC-9 Modified ReleaseAli UyPas encore d'évaluation
- Drug Induced Hepatitis: Dr.M.Sharmila Assistant Professor M7 (Prof CR Unit) Institute of Internal MedicineDocument21 pagesDrug Induced Hepatitis: Dr.M.Sharmila Assistant Professor M7 (Prof CR Unit) Institute of Internal MedicineAtakan Yeşil100% (1)
- Drug Absorption FarmakokinetikDocument15 pagesDrug Absorption FarmakokinetikiaafrianiPas encore d'évaluation
- Routes of Drug EliminationDocument17 pagesRoutes of Drug EliminationdahiphalehPas encore d'évaluation
- Pharmacology Practical Manual - Student Copy2Document11 pagesPharmacology Practical Manual - Student Copy2NareshPas encore d'évaluation
- Autacoid PharmacologyDocument38 pagesAutacoid PharmacologyAakkkPas encore d'évaluation
- Pharmacokinetics: Dr. Jahid MBBS, M.phil (Pharmacology) Head of Pharmacology (MD-AUCMS)Document52 pagesPharmacokinetics: Dr. Jahid MBBS, M.phil (Pharmacology) Head of Pharmacology (MD-AUCMS)vivianPas encore d'évaluation
- Nanoparticles in Cancer Therapy and DiagnosisDocument55 pagesNanoparticles in Cancer Therapy and Diagnosismimshin0% (1)
- Session 1 Introduction To PICS GMP 009-14Document12 pagesSession 1 Introduction To PICS GMP 009-14Elton SubijanoPas encore d'évaluation
- Design of Dosage FormDocument23 pagesDesign of Dosage FormIVORY DIANE AMANCIOPas encore d'évaluation
- Unit OintmentDocument39 pagesUnit OintmentEE KMPas encore d'évaluation
- Quinones: Prof. Dr. Ali Hikmet MeriçliDocument55 pagesQuinones: Prof. Dr. Ali Hikmet MeriçliEduard NiñoPas encore d'évaluation
- Drug Extraction RatioDocument8 pagesDrug Extraction RatioHeru Fajar SyaputraPas encore d'évaluation
- Nonlinear Pharmacokinetics 1Document11 pagesNonlinear Pharmacokinetics 1Mubashar ShahidPas encore d'évaluation
- Pharmaceutical Chemistry I (D. Pharm 1st Year)Document35 pagesPharmaceutical Chemistry I (D. Pharm 1st Year)PrathiPas encore d'évaluation
- OintmentDocument12 pagesOintmentErfaneh FNPas encore d'évaluation
- Katzung Chapter 24 Antiseizure DrugsDocument52 pagesKatzung Chapter 24 Antiseizure DrugsMeetAndreaPas encore d'évaluation
- BIOAVAILABILITY AND BIOEQUIVALANCE STUDIES Final - PPTX'Document32 pagesBIOAVAILABILITY AND BIOEQUIVALANCE STUDIES Final - PPTX'Md TayfuzzamanPas encore d'évaluation
- Pharmacokinetics CalculationDocument71 pagesPharmacokinetics CalculationBandameedi RamuPas encore d'évaluation
- BIOPHARMACEUTICS REDUCES BIOAVAILABILITY CONCERNSDocument38 pagesBIOPHARMACEUTICS REDUCES BIOAVAILABILITY CONCERNSDione TutanaPas encore d'évaluation
- Experiment 1: Preparation and Analysis of Laboratory BuffersDocument16 pagesExperiment 1: Preparation and Analysis of Laboratory BuffersMani ScoopsPas encore d'évaluation
- Pharmaceutical Buffers and Buffer CapacityDocument6 pagesPharmaceutical Buffers and Buffer Capacityharoon41Pas encore d'évaluation
- Pharmaceutical OrganizationDocument23 pagesPharmaceutical OrganizationPrecious ToledoPas encore d'évaluation
- PHARMA FABRIKON Company ProfileDocument17 pagesPHARMA FABRIKON Company Profilehonoureengomes3898Pas encore d'évaluation
- Hard HPMC CapsulesDocument3 pagesHard HPMC CapsulesnaturalcapsulesPas encore d'évaluation
- Handouts For Pharmaceutical formulations-ORIENTATION-18-19Document5 pagesHandouts For Pharmaceutical formulations-ORIENTATION-18-19Sarathchandran Chandrashekar ShenoyPas encore d'évaluation
- FDA Interpretation of 505 (B) (2) NDADocument36 pagesFDA Interpretation of 505 (B) (2) NDAShilpa KotianPas encore d'évaluation
- Master Formula RecordDocument39 pagesMaster Formula RecordAbhijit kanavaje100% (1)
- Data Farklin Apt. Faizal 27 Maret-18 April PDFDocument27 pagesData Farklin Apt. Faizal 27 Maret-18 April PDFfarklin rsuamiraPas encore d'évaluation
- Pharmcal Midterm DefDocument2 pagesPharmcal Midterm DefDakota SimbsPas encore d'évaluation
- TestDocument94 pagesTestmutiaraPas encore d'évaluation
- Compound Evaluation Form: Instructions For Preparation: CalculationsDocument3 pagesCompound Evaluation Form: Instructions For Preparation: CalculationsNeal AndersonPas encore d'évaluation
- AstyminDocument3 pagesAstyminNgozi GracePas encore d'évaluation
- 12-05-09 SOP Controlled Drugsreview V004Document18 pages12-05-09 SOP Controlled Drugsreview V004scribd-844801Pas encore d'évaluation
- ABDocument8 pagesABJia Weng FungPas encore d'évaluation
- PT. RUBEL ANUGERAH MEDICATAMA DAFTAR HARGADocument33 pagesPT. RUBEL ANUGERAH MEDICATAMA DAFTAR HARGAMalik TeamPas encore d'évaluation
- Pharma Cutoff HKDocument5 pagesPharma Cutoff HKAvinash GowdaPas encore d'évaluation
- Drug Interaction MonitoringDocument5 pagesDrug Interaction MonitoringAnonymousPas encore d'évaluation
- Anupama Cv-Pharmacist-2021Document2 pagesAnupama Cv-Pharmacist-2021ragesh r nairPas encore d'évaluation
- Pharmacology Exam COHORT 8Document9 pagesPharmacology Exam COHORT 8Lesley Liavoga SandePas encore d'évaluation
- Pharmaceutical JurisprudenceDocument2 pagesPharmaceutical JurisprudenceMasum Billa MollaPas encore d'évaluation
- INORGANIC COLLOIDSDocument1 pageINORGANIC COLLOIDSAryasingh5656Pas encore d'évaluation
- Patient CounselingDocument3 pagesPatient CounselingmrkrlndPas encore d'évaluation
- Expired DrugsDocument4 pagesExpired DrugsWulan Gatra100% (1)
- Common Acronyms & AbbreviationsDocument2 pagesCommon Acronyms & AbbreviationsMayrigen DominguezPas encore d'évaluation
- PNF AssignmentDocument2 pagesPNF AssignmentCamila BarzagaPas encore d'évaluation
- 1 Early Access Application 14 May 2018 PDFDocument2 pages1 Early Access Application 14 May 2018 PDFArdat LiliPas encore d'évaluation
- Monitoring Penggunaan Obat Generik di Rumah Sakit Ciputra HospitalDocument15 pagesMonitoring Penggunaan Obat Generik di Rumah Sakit Ciputra HospitalMujiyanto RichardPas encore d'évaluation
- Materi Ibu Difa - Pelayanan ResepDocument18 pagesMateri Ibu Difa - Pelayanan ResepCang HaedarPas encore d'évaluation
- ACTD Format Part IDocument14 pagesACTD Format Part Iultimate_2226252Pas encore d'évaluation
- Oral Medication AdministrationDocument13 pagesOral Medication AdministrationDylan HimoPas encore d'évaluation
- Chapter 26: Prescribing Medications in Pediatrics Garzon Maaks: Burns' Pediatric Primary Care, 7th EditionDocument3 pagesChapter 26: Prescribing Medications in Pediatrics Garzon Maaks: Burns' Pediatric Primary Care, 7th EditionHelen UgochukwuPas encore d'évaluation