Vous aimerez peut-être aussi
- Protein MetabolismDocument78 pagesProtein MetabolismU2002862 STUDENTPas encore d'évaluation
- Amino Acids and ProteinsDocument56 pagesAmino Acids and Proteinstahaniu gouaouPas encore d'évaluation
- Protein ChemistryDocument35 pagesProtein Chemistryمريم البندرPas encore d'évaluation
- Nutrition 1Document8 pagesNutrition 1Marwan AlaaPas encore d'évaluation
- asset-v1_HKVU+COSAS+2021_Q4_R1+type@asset+block@metal-seminarDocument37 pagesasset-v1_HKVU+COSAS+2021_Q4_R1+type@asset+block@metal-seminar1126playpubgPas encore d'évaluation
- UreacycleDocument18 pagesUreacycleChudasama DhruvrajsinhPas encore d'évaluation
- Enzymatic catalysis and protein structure and functionDocument88 pagesEnzymatic catalysis and protein structure and functionRazvan MihaiPas encore d'évaluation
- LecturerDocument29 pagesLectureratef.salman.grPas encore d'évaluation
- Amines: - Organic Compounds of Nitrogen N - Classified As Primary, Secondary, TertiaryDocument44 pagesAmines: - Organic Compounds of Nitrogen N - Classified As Primary, Secondary, Tertiaryshah_jalpan92911Pas encore d'évaluation
- Lecture 4Document31 pagesLecture 4anaPas encore d'évaluation
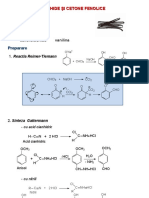
- Aldehide Şi Cetone FenoliceDocument9 pagesAldehide Şi Cetone FenoliceMarinelaPas encore d'évaluation
- Amino Acid Metabolism: Hanley N. Abramson Professor of Pharmaceutical Sciences Wayne State University December 2009Document51 pagesAmino Acid Metabolism: Hanley N. Abramson Professor of Pharmaceutical Sciences Wayne State University December 2009vigsaranPas encore d'évaluation
- Acid Carboxylic Beùo Vaø Daãn ChaátDocument38 pagesAcid Carboxylic Beùo Vaø Daãn ChaátthidaithanhPas encore d'évaluation
- Acid Carboxylic Beùo Vaø Daãn ChaátDocument38 pagesAcid Carboxylic Beùo Vaø Daãn ChaátBích Nguyễn NgọcPas encore d'évaluation
- AAs&ProteinsDocument33 pagesAAs&ProteinsMonica BañasPas encore d'évaluation
- Nutrición Proteínas en InglesDocument16 pagesNutrición Proteínas en InglesJennifer RamosPas encore d'évaluation
- Asam Amino OkDocument31 pagesAsam Amino OkYuniar YuniarPas encore d'évaluation
- Formula of The 20 Common Amino AcidsDocument11 pagesFormula of The 20 Common Amino AcidsAngelica AngelesPas encore d'évaluation
- Uii10g18 10 2023 09 02 44Document10 pagesUii10g18 10 2023 09 02 44asoom 111Pas encore d'évaluation
- Amino Acid Metabolism: Susi Endrini, PH.DDocument15 pagesAmino Acid Metabolism: Susi Endrini, PH.DRA viPas encore d'évaluation
- Protein Structure and FucntionsDocument59 pagesProtein Structure and FucntionsTiffany Shane VallentePas encore d'évaluation
- Amino Acids ProteinsDocument30 pagesAmino Acids ProteinsLoh JiayeePas encore d'évaluation
- EAMCET QR Chemistry SR Chem 17.organic Chemistry Carboxylic AcidsDocument5 pagesEAMCET QR Chemistry SR Chem 17.organic Chemistry Carboxylic AcidsJagadeesh Goli100% (2)
- 11.alcohol, Phenol & Ethers Colour BookletDocument84 pages11.alcohol, Phenol & Ethers Colour BookletVishal Malik100% (1)
- Proteins Part I: Amino Acids and PeptidesDocument62 pagesProteins Part I: Amino Acids and PeptidesDaniele Joseph HizonPas encore d'évaluation
- 04 Amino AcidsDocument42 pages04 Amino AcidsAbdo MohdyPas encore d'évaluation
- Chemical RXN - Aldehydes, KetoneDocument11 pagesChemical RXN - Aldehydes, Ketoneudhayadeepak60Pas encore d'évaluation
- Amino Acid Structure and PropertiesDocument22 pagesAmino Acid Structure and PropertiesHiroPas encore d'évaluation
- Amino Acids Metabolism &: Urea CycleDocument11 pagesAmino Acids Metabolism &: Urea Cyclemuhammad amjadPas encore d'évaluation
- Amino Acids and Peptide BondDocument61 pagesAmino Acids and Peptide Bondranger cleetusPas encore d'évaluation
- Reactions and Preparations of Aldehydes and KetonesDocument5 pagesReactions and Preparations of Aldehydes and KetonesJAN JERICHO MENTOYPas encore d'évaluation
- Non Polar-Hydrophobic-Buried in Protein Core-AliphaticDocument4 pagesNon Polar-Hydrophobic-Buried in Protein Core-AliphaticRob FranciscoPas encore d'évaluation
- Organic Chemistry 2 - Chem - f3 - v1Document66 pagesOrganic Chemistry 2 - Chem - f3 - v1Lubanga N JamesPas encore d'évaluation
- ReactiiDocument8 pagesReactiiEddyPas encore d'évaluation
- CHE 311 Lecture 1 - Peptide Structure DeterminationDocument57 pagesCHE 311 Lecture 1 - Peptide Structure Determinationisaac mwanzaPas encore d'évaluation
- AMINOKISELINE HEMIJSKE OSOBINEDocument42 pagesAMINOKISELINE HEMIJSKE OSOBINEErK050% (2)
- NMR_controlled_titrationDocument162 pagesNMR_controlled_titrationSamuel AguiarPas encore d'évaluation
- 11.alcohol, Phenol & Ethers Colour Booklet PDFDocument59 pages11.alcohol, Phenol & Ethers Colour Booklet PDFMridu BhandariPas encore d'évaluation
- PROTEIN ANALYSIS AND APPLICATIONSDocument52 pagesPROTEIN ANALYSIS AND APPLICATIONSMUHAMMAD ADLANPas encore d'évaluation
- Organic Chemistry Reactions and NomenclatureDocument20 pagesOrganic Chemistry Reactions and NomenclatureCoolz TricksPas encore d'évaluation
- Organic Chemistry I: Ganesha University of EducationDocument21 pagesOrganic Chemistry I: Ganesha University of EducationUnii ChemPas encore d'évaluation
- Structural Formulas of Organic CompoundsDocument20 pagesStructural Formulas of Organic Compoundstarchettifrancisco0% (1)
- Amino Acids and PeptidesDocument38 pagesAmino Acids and PeptidesrizwanamalikPas encore d'évaluation
- Chemistry Online NotesDocument16 pagesChemistry Online NotesBharti YadavPas encore d'évaluation
- Chemistry in Everyday LifeDocument4 pagesChemistry in Everyday Lifevdonga9Pas encore d'évaluation
- AEMGP 9 - Mutu Protein Pangan IDocument21 pagesAEMGP 9 - Mutu Protein Pangan IJulya GHPas encore d'évaluation
- Biosíntesis de Purinas y PirimidinasDocument18 pagesBiosíntesis de Purinas y PirimidinasXimenaPas encore d'évaluation
- Figures ScratchDocument7 pagesFigures ScratchJerryson OrpillaPas encore d'évaluation
- Adobe Scan 30-Aug-2022Document5 pagesAdobe Scan 30-Aug-2022Harshit RajPas encore d'évaluation
- Amino Acid TableDocument1 pageAmino Acid TableEugene OkpanteyPas encore d'évaluation
- Biochemistry for Allied Medical Courses: Proteins and Amino AcidsDocument50 pagesBiochemistry for Allied Medical Courses: Proteins and Amino AcidsFelica Delos ReyesPas encore d'évaluation
- Macam-Macam Asam AminoDocument2 pagesMacam-Macam Asam Aminoarif nur rokhmanPas encore d'évaluation
- 5lkmq-Acides AminesDocument1 page5lkmq-Acides Aminessoumailakaila59Pas encore d'évaluation
- Amines: Subjective ProblemsDocument7 pagesAmines: Subjective ProblemsBharti SharmaPas encore d'évaluation
- Alkanes: 1.1 Classification of HydrocarbonDocument33 pagesAlkanes: 1.1 Classification of HydrocarbonKhizra TehreemPas encore d'évaluation
- Classification of Org. CompdDocument7 pagesClassification of Org. CompdRaju SinghPas encore d'évaluation
- AGEs and the Kidney: Role in Pathogenesis of Vascular and Renal DiseaseDocument14 pagesAGEs and the Kidney: Role in Pathogenesis of Vascular and Renal DiseaseKarina ThiemePas encore d'évaluation
- Série 2 CHIMIEDocument3 pagesSérie 2 CHIMIERim AbouttiPas encore d'évaluation
- Solution Manual for The Elements of Polymer Science and EngineeringD'EverandSolution Manual for The Elements of Polymer Science and EngineeringÉvaluation : 4 sur 5 étoiles4/5 (3)
- CH—Acids: A Guide to All Existing Problems of CH-Acidity with New Experimental Methods and Data, Including Indirect Electrochemical, Kinetic and Thermodynamic StudiesD'EverandCH—Acids: A Guide to All Existing Problems of CH-Acidity with New Experimental Methods and Data, Including Indirect Electrochemical, Kinetic and Thermodynamic StudiesPas encore d'évaluation
- Uas Inggris FixDocument13 pagesUas Inggris FixIbi Yulia SetyaniPas encore d'évaluation
- OjldDocument17 pagesOjldIbi Yulia SetyaniPas encore d'évaluation
- Resume of Respiratory ProblemDocument5 pagesResume of Respiratory ProblemIbi Yulia SetyaniPas encore d'évaluation
- LP EliminasiDocument1 pageLP EliminasiIbi Yulia SetyaniPas encore d'évaluation
- Teaching ReadingDocument5 pagesTeaching ReadingIbi Yulia SetyaniPas encore d'évaluation
- Proposal GreetingDocument8 pagesProposal GreetingIbi Yulia SetyaniPas encore d'évaluation
- Implementasi CKD SeminarDocument14 pagesImplementasi CKD SeminarIbi Yulia SetyaniPas encore d'évaluation
- Teaching ReadingDocument5 pagesTeaching ReadingIbi Yulia SetyaniPas encore d'évaluation
- Implementasi CKD SeminarDocument14 pagesImplementasi CKD SeminarIbi Yulia SetyaniPas encore d'évaluation
- Implementasi CKD SeminarDocument14 pagesImplementasi CKD SeminarIbi Yulia SetyaniPas encore d'évaluation
- Resume of Respiratory ProblemDocument5 pagesResume of Respiratory ProblemIbi Yulia SetyaniPas encore d'évaluation
- Resume of Respiratory ProblemDocument5 pagesResume of Respiratory ProblemIbi Yulia SetyaniPas encore d'évaluation
- 9-Reproductive HealthDocument32 pages9-Reproductive HealthLiezel CauilanPas encore d'évaluation
- Resume AfidDocument5 pagesResume AfidIbi Yulia SetyaniPas encore d'évaluation
- Pidato Jokowi Apec Ceo Summit 2014Document3 pagesPidato Jokowi Apec Ceo Summit 2014Ibi Yulia SetyaniPas encore d'évaluation
- Respiratory Exam GuideDocument18 pagesRespiratory Exam GuideIbi Yulia Setyani100% (1)
- Proposal GreetingDocument8 pagesProposal GreetingIbi Yulia SetyaniPas encore d'évaluation
- LP EliminasiDocument1 pageLP EliminasiIbi Yulia SetyaniPas encore d'évaluation
- Reproductive System: Nursing AssessmentDocument28 pagesReproductive System: Nursing AssessmentIbi Yulia SetyaniPas encore d'évaluation
- Komunikasi, Jurnal Roro Kelompok 3Document14 pagesKomunikasi, Jurnal Roro Kelompok 3Ibi Yulia SetyaniPas encore d'évaluation
- 9-Reproductive HealthDocument32 pages9-Reproductive HealthLiezel CauilanPas encore d'évaluation
- Understand Computer Terms CorrectlyDocument5 pagesUnderstand Computer Terms CorrectlyIbi Yulia SetyaniPas encore d'évaluation
- Introduction To Reproductive Health and The Environment: (Draft For Review)Document70 pagesIntroduction To Reproductive Health and The Environment: (Draft For Review)Ibi Yulia SetyaniPas encore d'évaluation
- 9-Reproductive HealthDocument32 pages9-Reproductive HealthLiezel CauilanPas encore d'évaluation
- Respiratory Exam GuideDocument18 pagesRespiratory Exam GuideIbi Yulia Setyani100% (1)
- Kep Anak - PDK EngDocument18 pagesKep Anak - PDK EngIbi Yulia SetyaniPas encore d'évaluation
- Protein MetabolismDocument37 pagesProtein MetabolismIbi Yulia SetyaniPas encore d'évaluation
- Nursing DocumentationDocument7 pagesNursing DocumentationIbi Yulia SetyaniPas encore d'évaluation
- 192 Nilai 2Document13 pages192 Nilai 2Ibi Yulia SetyaniPas encore d'évaluation
- CBC and CRP ReportDocument3 pagesCBC and CRP Reportrajasereddy1275Pas encore d'évaluation
- Inno-Lia HCV ScoreDocument88 pagesInno-Lia HCV ScoreKatherine OhaPas encore d'évaluation
- Lec-1 BLOOD - 2 (Cardiac Physiology)Document20 pagesLec-1 BLOOD - 2 (Cardiac Physiology)Wilson CheungPas encore d'évaluation
- Mc3 Chapter 1Document7 pagesMc3 Chapter 1Lorraine RiegoPas encore d'évaluation
- Blood BookletDocument10 pagesBlood BookletTezdjan HassanPas encore d'évaluation
- Central Dogma of Molecular BiologyDocument7 pagesCentral Dogma of Molecular BiologyRenee Louise CasullaPas encore d'évaluation
- Caiazza, Toole - 2003 PDFDocument4 pagesCaiazza, Toole - 2003 PDFNICOLÁS SEBASTÍAN GOMEZ SEQUEDAPas encore d'évaluation
- American Society of Hematology Self-Assessment Program (Adam Cuker, Jessica K. Altman, Aaron T. Gerds Etc.)Document772 pagesAmerican Society of Hematology Self-Assessment Program (Adam Cuker, Jessica K. Altman, Aaron T. Gerds Etc.)Luciano LaranjeiraPas encore d'évaluation
- Week 9 Lab Exercise - Immune SystemDocument3 pagesWeek 9 Lab Exercise - Immune SystemJoselito JardielPas encore d'évaluation
- What Are mRNA VaccinesDocument4 pagesWhat Are mRNA VaccinesGabriela Trujillo Salcedo50% (2)
- Skin Exposure To Narrow Band Ultraviolet (UVB) Light Modulates The Human Intestinal MicrobiomeDocument11 pagesSkin Exposure To Narrow Band Ultraviolet (UVB) Light Modulates The Human Intestinal MicrobiomesimasPas encore d'évaluation
- The Biology Project - Meiosis TutorialDocument8 pagesThe Biology Project - Meiosis TutorialJimmie MaddoxPas encore d'évaluation
- Animal Bite Module 2021Document20 pagesAnimal Bite Module 2021ERMED OFC R2TMCPas encore d'évaluation
- European Foulbrood of Honey Bees: 1. Identification of The AgentDocument5 pagesEuropean Foulbrood of Honey Bees: 1. Identification of The AgentPınar BarkanPas encore d'évaluation
- MalariaDocument21 pagesMalariayusak tapakedingPas encore d'évaluation
- Jim Stone Freelance 4-13-2021Document96 pagesJim Stone Freelance 4-13-2021Brian CharlesPas encore d'évaluation
- Medical ProtozoologyDocument6 pagesMedical ProtozoologyRaymund MontoyaPas encore d'évaluation
- Haematinics, Coagulants and Anticoagulant - DR - Jibachha Sah, M.V.SC, LecturerDocument27 pagesHaematinics, Coagulants and Anticoagulant - DR - Jibachha Sah, M.V.SC, Lecturerjibachha sahPas encore d'évaluation
- Import Routes and Nuclear Functions of Argonaute and Other Small RNA - Silencing ProteinsDocument12 pagesImport Routes and Nuclear Functions of Argonaute and Other Small RNA - Silencing ProteinsMariaAndreaLaraSalasPas encore d'évaluation
- Enteric Fever .HarrisonDocument7 pagesEnteric Fever .Harrisonqayyum consultantfpscPas encore d'évaluation
- Cavallieri Et Al (2023 Cells) PD DJ-1 RevDocument17 pagesCavallieri Et Al (2023 Cells) PD DJ-1 RevKrisztina TothPas encore d'évaluation
- AIIMS 2019 Biology Sample Question PaperDocument10 pagesAIIMS 2019 Biology Sample Question PapermisostudyPas encore d'évaluation
- BIO X ICSE GeneticsDocument13 pagesBIO X ICSE Geneticsrkjoseph1410Pas encore d'évaluation
- Msi IhcDocument30 pagesMsi IhcAnkush NayyarPas encore d'évaluation
- Process and Molecular Modelling Study of Entecavir Drug-Resistant HBVDocument9 pagesProcess and Molecular Modelling Study of Entecavir Drug-Resistant HBVDrkrishnasarma pathyPas encore d'évaluation
- Lecture Notes Antibody Structure & FunctionDocument5 pagesLecture Notes Antibody Structure & FunctionNuti FafaPas encore d'évaluation
- Nucleic Acid Extraction MethodsDocument25 pagesNucleic Acid Extraction Methodsmasthan6yPas encore d'évaluation
- Soyfoods and Soybean Products: From Traditional Use To Modern ApplicationsDocument15 pagesSoyfoods and Soybean Products: From Traditional Use To Modern ApplicationsaudaceramPas encore d'évaluation
- AIDS and Ebola: WeaponizationDocument14 pagesAIDS and Ebola: Weaponizationdavid0217100% (1)
- Mtap - Virology NotesDocument7 pagesMtap - Virology NotesMoira Pauline LibroraniaPas encore d'évaluation