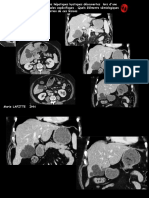
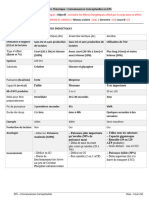
Vous aimerez peut-être aussi
- Paul MorayDocument14 pagesPaul MorayNini TotoPas encore d'évaluation
- Elements D'hemostaseDocument10 pagesElements D'hemostasegjobPas encore d'évaluation
- Tpe Elisa DirectDocument11 pagesTpe Elisa DirectgjobPas encore d'évaluation
- Techniques en ImmunologieDocument44 pagesTechniques en Immunologiegjob75% (4)
- Cours de ParasitologieDocument24 pagesCours de Parasitologiegjob100% (1)
- Invitation Biomr SimemDocument4 pagesInvitation Biomr SimemKhadidja BensmainePas encore d'évaluation
- ExercicesDocument44 pagesExercicesmanal treegreen100% (1)
- Tdiettdii: Nadh Nadh - 1 - 1 Nadh - 1 - 1Document6 pagesTdiettdii: Nadh Nadh - 1 - 1 Nadh - 1 - 1youssefbgh03Pas encore d'évaluation
- Examen Blanc PD Vogt Sequence Six 2021 - Copie - Copie - Copie - CopieDocument6 pagesExamen Blanc PD Vogt Sequence Six 2021 - Copie - Copie - Copie - Copiecyrille nlendPas encore d'évaluation
- Module 5 Pre Paration Et Techniques de Magne TismeDocument33 pagesModule 5 Pre Paration Et Techniques de Magne TismenathaliebariselePas encore d'évaluation
- GlucidesDocument34 pagesGlucidesRémi Cechia100% (1)
- Proceeding Congres Microbiod-2Document245 pagesProceeding Congres Microbiod-2matmat yasminePas encore d'évaluation
- BiochimieDocument36 pagesBiochimieurdorous - يور دروسPas encore d'évaluation
- Benahmed PDFDocument10 pagesBenahmed PDFoussama22Pas encore d'évaluation
- La Preparation Physique Integree N0ig6zDocument60 pagesLa Preparation Physique Integree N0ig6zJulien MunozPas encore d'évaluation
- CR 5 - DPNDocument89 pagesCR 5 - DPNMouad HiliaPas encore d'évaluation
- Les JumeauxDocument30 pagesLes JumeauxDją ŻıraPas encore d'évaluation
- Td2 Corrige TypeDocument3 pagesTd2 Corrige TypeAsmae EssarhiPas encore d'évaluation
- Le Compte 1958Document18 pagesLe Compte 1958Anonymous 6iBKWPWPas encore d'évaluation
- cystadenome-biliaire-ML-2012Document23 pagescystadenome-biliaire-ML-2012Jerome Gluck DelannoyPas encore d'évaluation
- DRS5 Races-Classes 6Document13 pagesDRS5 Races-Classes 6TheurelPas encore d'évaluation
- Présentations Céphaliques Défléchies Ayoub YaiciDocument6 pagesPrésentations Céphaliques Défléchies Ayoub YaiciAmina AmiinaPas encore d'évaluation
- Chapitre 2 Dernière Version Biologie Cellulaire DR LAMDA 2021-2022Document108 pagesChapitre 2 Dernière Version Biologie Cellulaire DR LAMDA 2021-2022Zakaria BbaPas encore d'évaluation
- Tendon ConjointDocument41 pagesTendon ConjointElbordjiPas encore d'évaluation
- TB TP 3 3 Meiose Brassage GenetiqueDocument16 pagesTB TP 3 3 Meiose Brassage GenetiqueOmar sabirPas encore d'évaluation
- TP 1Document7 pagesTP 1Racha DouniaPas encore d'évaluation
- Beispiel B2 CorrecteurDocument6 pagesBeispiel B2 CorrecteurMustapha Mektan0% (1)
- EPS - Cours Théorique 2bacDocument1 pageEPS - Cours Théorique 2bacahlame elallaliPas encore d'évaluation
- Composition Chimique Des Coproduits de La ViandeDocument3 pagesComposition Chimique Des Coproduits de La ViandeDahlia SanouPas encore d'évaluation