Vous aimerez peut-être aussi
- Équations différentielles: Les Grands Articles d'UniversalisD'EverandÉquations différentielles: Les Grands Articles d'UniversalisPas encore d'évaluation
- Algebre Lineaire Et Geometrie Des Polyno PDFDocument11 pagesAlgebre Lineaire Et Geometrie Des Polyno PDFSat KoosPas encore d'évaluation
- CoursDocument8 pagesCoursfrédéric MasialaPas encore d'évaluation
- Series de FourierDocument9 pagesSeries de FourierTizirii AdouamaPas encore d'évaluation
- Agreg FourierDocument19 pagesAgreg FourierYOUSSEF TAIKPas encore d'évaluation
- Exam Meca An Janvier 2012 PDFDocument4 pagesExam Meca An Janvier 2012 PDFmadani abdelhamidPas encore d'évaluation
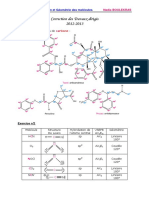
- Correction 2Document11 pagesCorrection 2Marcelle BillongPas encore d'évaluation
- Analyse Des Matrice Aleatoire IntroductionDocument143 pagesAnalyse Des Matrice Aleatoire Introductionzain zagoraPas encore d'évaluation
- Banque X-ÉNS 2020 MP Mathématiques B EaDocument3 pagesBanque X-ÉNS 2020 MP Mathématiques B EaMehdi BnmssdPas encore d'évaluation
- Mines 2007 SerieDocument4 pagesMines 2007 SerieAmine ZitounPas encore d'évaluation
- DS3 1Document4 pagesDS3 1Yahya El GueddariPas encore d'évaluation
- Exochapitre 0 CorrDocument14 pagesExochapitre 0 Corranouchkantsika9Pas encore d'évaluation
- MinesPonts 2007 MP M1 EnonceDocument4 pagesMinesPonts 2007 MP M1 Enoncesusu XXPas encore d'évaluation
- ds06 1718Document4 pagesds06 1718Saley NouroudinePas encore d'évaluation
- MP Maths X 2 2011.enonceDocument4 pagesMP Maths X 2 2011.enoncepepafov154moneyzonPas encore d'évaluation
- TD2 21 22Document3 pagesTD2 21 22Chiha WissemPas encore d'évaluation
- Mec Analytique Et Vibration Examens 01Document2 pagesMec Analytique Et Vibration Examens 01Hamza El-watiqPas encore d'évaluation
- Exam Janvier 10Document2 pagesExam Janvier 10adilbourejylahPas encore d'évaluation
- Fonctions Speciales Et Polynomes Orthogonaux Cour-75-96Document22 pagesFonctions Speciales Et Polynomes Orthogonaux Cour-75-96ghouti ghoutiPas encore d'évaluation
- Decembre 2016Document3 pagesDecembre 2016Doha FriatPas encore d'évaluation
- CorrigeExamen17 18 BerradaDocument4 pagesCorrigeExamen17 18 BerradaAwatif BePas encore d'évaluation
- Corrige Phys Stat TD1Document10 pagesCorrige Phys Stat TD1mariem OuriPas encore d'évaluation
- Exo Geo DiffDocument16 pagesExo Geo DiffGEOFFROY AIHOUPas encore d'évaluation
- Autonomie2 - 20202021 CorrectionDocument3 pagesAutonomie2 - 20202021 CorrectionGhada AmakranePas encore d'évaluation
- corrigeMG S1 1920Document5 pagescorrigeMG S1 1920Hamza SadikPas encore d'évaluation
- 16 EuclidexoDocument5 pages16 EuclidexohmzbhiproPas encore d'évaluation
- M 213 M 2 EcDocument4 pagesM 213 M 2 EcLM --Pas encore d'évaluation
- Cochran FisherDocument2 pagesCochran FisherHad Candide CODJOPas encore d'évaluation
- Oblique EnonceDocument3 pagesOblique EnoncemouhcinePas encore d'évaluation
- Cyclotomie TD CorrectionDocument12 pagesCyclotomie TD Correctionrayan ben saidPas encore d'évaluation
- Cours Simulation Alea To I ReDocument57 pagesCours Simulation Alea To I ReAdji DioufPas encore d'évaluation
- Feuille 3Document4 pagesFeuille 3Hugo paléoPas encore d'évaluation
- Centrale 2016 - PSI 2 Un Corrig e 1 Transformation de FourierDocument11 pagesCentrale 2016 - PSI 2 Un Corrig e 1 Transformation de FourierAlexandre LamPas encore d'évaluation
- 32 Dimension Planche CorrigesDocument8 pages32 Dimension Planche CorrigesIbrahimPas encore d'évaluation
- Exo Fourier 8Document4 pagesExo Fourier 8medPas encore d'évaluation
- Repet 2Document5 pagesRepet 2redwanePas encore d'évaluation
- Devoir Polynomes OrthogonauxDocument2 pagesDevoir Polynomes OrthogonauxcristianoronadomarocainPas encore d'évaluation
- Mecanique Quantique Chapitre 4: Solutions Stationnaires de L' Equation de SCHR OdingerDocument18 pagesMecanique Quantique Chapitre 4: Solutions Stationnaires de L' Equation de SCHR OdingerAnwar MoudadPas encore d'évaluation
- MyPrepa PolynomesDocument20 pagesMyPrepa PolynomesElfeur NGOLOPas encore d'évaluation
- LM345 TD5solDocument6 pagesLM345 TD5solkukis14Pas encore d'évaluation
- 5f564404bfb2cITS - B - Maths - 2020 - SujetsDocument12 pages5f564404bfb2cITS - B - Maths - 2020 - Sujetstamba vieux tolnoPas encore d'évaluation
- 2021 2022 Pcsi EtudefrequentielleDocument26 pages2021 2022 Pcsi EtudefrequentielleKhalid ElmentagiPas encore d'évaluation
- Chapitre1 2Document36 pagesChapitre1 2Mohammed HachemiPas encore d'évaluation
- QCM Outils Maths 2022Document4 pagesQCM Outils Maths 2022ikr3oukPas encore d'évaluation
- MinesPonts 2023 PC M1 EnonceDocument6 pagesMinesPonts 2023 PC M1 Enonceone piece to drawPas encore d'évaluation
- Grégory Berhuy - Suites Et Séries de Fonctions (Lecture Notes) (2015)Document88 pagesGrégory Berhuy - Suites Et Séries de Fonctions (Lecture Notes) (2015)Chatiri AzizPas encore d'évaluation
- Mec Analytique Et Vibration Examens 01 - CompressedDocument2 pagesMec Analytique Et Vibration Examens 01 - Compressedbokpedidier1Pas encore d'évaluation
- Centrale PSI 1 - 2021cDocument4 pagesCentrale PSI 1 - 2021cAdrien GalezowskiPas encore d'évaluation
- Condition de RaccordementDocument33 pagesCondition de RaccordementMehdi El HamdouchiPas encore d'évaluation
- Exercices L12Document19 pagesExercices L12Alassane SowPas encore d'évaluation
- VirielDocument8 pagesVirielImen SfarPas encore d'évaluation
- ds09 Un Endomorphisme SujetDocument4 pagesds09 Un Endomorphisme Sujetanis.benh05Pas encore d'évaluation
- Cor3 Tore Proj SphereDocument9 pagesCor3 Tore Proj SphereBey LpPas encore d'évaluation
- PolynomesTchebychev PDFDocument11 pagesPolynomesTchebychev PDFMalick DiopPas encore d'évaluation
- Devoir ' A La Maison N 8Document1 pageDevoir ' A La Maison N 8mehdi benmassoudPas encore d'évaluation
- 1 Matrice de Transition Et Cha Ines de MarkovDocument6 pages1 Matrice de Transition Et Cha Ines de MarkovCyrille LamasséPas encore d'évaluation
- DM10 Bis (Mines-Centrale Nombre Dyadiques)Document4 pagesDM10 Bis (Mines-Centrale Nombre Dyadiques)S cihikPas encore d'évaluation
- EuclidienDocument8 pagesEuclidienJean HurtePas encore d'évaluation
- PC MATHS X 1 2014.enonceDocument6 pagesPC MATHS X 1 2014.enonceBile DjetouanPas encore d'évaluation
- M1edp CorrigesDocument83 pagesM1edp CorrigesJustin EssonoPas encore d'évaluation
- 2018-2019 IE1 CorrigéDocument2 pages2018-2019 IE1 CorrigéEugene FoucherPas encore d'évaluation
- Figures-Liaisons ChimiquesDocument45 pagesFigures-Liaisons ChimiquesKoùukoù AbbàssiiPas encore d'évaluation
- La Liaison ChimiqueDocument39 pagesLa Liaison ChimiqueSophie Hamon100% (1)
- 2 Hybridation Et Geometrie Des Molecules 1 PDFDocument19 pages2 Hybridation Et Geometrie Des Molecules 1 PDFMINDANOU SHEIKH ALIOU DJAGNEPas encore d'évaluation
- Cours Liaison Chimique - CovalenteDocument78 pagesCours Liaison Chimique - Covalentemanalch1219Pas encore d'évaluation
- QCM Corrige - VesprDocument14 pagesQCM Corrige - VesprAbdelOuahidSenhadji100% (3)
- Chapitre VII: 1. Principe Des Calculs DFTDocument12 pagesChapitre VII: 1. Principe Des Calculs DFTMoha Med100% (1)
- Cours de Liaison Chimique EtudiantsDocument50 pagesCours de Liaison Chimique EtudiantsAnge PastoréPas encore d'évaluation
- Solution TD 3Document6 pagesSolution TD 3Rhm GamingPas encore d'évaluation
- Chapitre 05 Liaisons ChimiquesDocument17 pagesChapitre 05 Liaisons Chimiquesامين لتصميم الديكوراتPas encore d'évaluation
- CC 2 CorrigeDocument4 pagesCC 2 CorrigeRuego RuegoSgPas encore d'évaluation
- Concept D'Hybridation D'Orbitales AtomiquesDocument8 pagesConcept D'Hybridation D'Orbitales AtomiquesnonoooPas encore d'évaluation
- Cours Modelisation MoleculaireDocument16 pagesCours Modelisation MoleculaireKGGKFPas encore d'évaluation
- Hartree FockDocument18 pagesHartree FockKhalid ZegPas encore d'évaluation
- Cours Liaisons Chimiques 20-20Document57 pagesCours Liaisons Chimiques 20-20ussamautshihaPas encore d'évaluation
- ChmTheo S5 Chapitre 7Document32 pagesChmTheo S5 Chapitre 7Abdelhakim BailalPas encore d'évaluation
- TP LIAISONS MOLCAL 2022-2023 - MoctarDocument10 pagesTP LIAISONS MOLCAL 2022-2023 - Moctarmactar sarrPas encore d'évaluation
- Amphi 2Document40 pagesAmphi 2api-3830967Pas encore d'évaluation
- TD Orbitalaire L3 2019-1Document31 pagesTD Orbitalaire L3 2019-1dhoubzainabPas encore d'évaluation
- Rapport de Chimie ComputationnelleDocument24 pagesRapport de Chimie ComputationnelleErick basiluaPas encore d'évaluation
- Cours Chimie Orga Hybridation PDFDocument18 pagesCours Chimie Orga Hybridation PDFniniPas encore d'évaluation
- TD Hybridation 6 SolutionDocument2 pagesTD Hybridation 6 SolutionLoïc ITOUAPas encore d'évaluation
- Hückel Elèves TDDocument3 pagesHückel Elèves TDami rPas encore d'évaluation
- Chap M6Document88 pagesChap M6Kader MilanoPas encore d'évaluation
- Compte Rendu TP 1: Modélisation Moléculaire Séance Dirigée Sur Ordinateur: Méthode de Huckel SimpleDocument12 pagesCompte Rendu TP 1: Modélisation Moléculaire Séance Dirigée Sur Ordinateur: Méthode de Huckel SimpleyousralahlebPas encore d'évaluation
- Controle 2020 QuantiqueDocument2 pagesControle 2020 QuantiqueRhm Gaming100% (1)
- Fichier Produit 2114Document52 pagesFichier Produit 2114FlorinaPas encore d'évaluation
- Chapitre 2Document39 pagesChapitre 2Vera Legba-MonyPas encore d'évaluation
- Chapitre 4Document9 pagesChapitre 4LkhaybPas encore d'évaluation
- Methode de Huckel - RevisionsDocument6 pagesMethode de Huckel - RevisionsAyyoub DahbiPas encore d'évaluation
- Améliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesD'EverandAméliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesÉvaluation : 5 sur 5 étoiles5/5 (2)
- Technologie automobile: Les Grands Articles d'UniversalisD'EverandTechnologie automobile: Les Grands Articles d'UniversalisPas encore d'évaluation
- Je me prépare aux examens du ministère en mathématiques: Es-tu prêt à passer le test ?D'EverandJe me prépare aux examens du ministère en mathématiques: Es-tu prêt à passer le test ?Évaluation : 4 sur 5 étoiles4/5 (1)
- Électrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsD'EverandÉlectrotechnique | Pas à Pas: Bases, composants & circuits expliqués pour les débutantsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Revue des incompris revue d'histoire des oubliettes: Le Réveil de l'Horloge de Célestin Louis Maxime Dubuisson aliéniste et poèteD'EverandRevue des incompris revue d'histoire des oubliettes: Le Réveil de l'Horloge de Célestin Louis Maxime Dubuisson aliéniste et poèteÉvaluation : 3 sur 5 étoiles3/5 (3)
- Anatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsD'EverandAnatomie & 100 étirements essentiels pour le running: Principes de base, Techniques, Tableaux de séries, Précautions à prendre, Conseils, Programmes d'étirementsPas encore d'évaluation
- Manuel pour les débutants Fabriquez des savons naturelsD'EverandManuel pour les débutants Fabriquez des savons naturelsÉvaluation : 3 sur 5 étoiles3/5 (2)
- Harmonisation Energétique des Personnes: Manuel de Curothérapie 2020D'EverandHarmonisation Energétique des Personnes: Manuel de Curothérapie 2020Évaluation : 4 sur 5 étoiles4/5 (8)
- Géobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainD'EverandGéobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)
- Affirmations positives : Perte de poids pour les femmesD'EverandAffirmations positives : Perte de poids pour les femmesPas encore d'évaluation
- Vous saurez tout sur le permis: Un livre rassurant pour les maudits du volantD'EverandVous saurez tout sur le permis: Un livre rassurant pour les maudits du volantPas encore d'évaluation
- 20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsD'Everand20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Production et propagation des sons: Les Grands Articles d'UniversalisD'EverandProduction et propagation des sons: Les Grands Articles d'UniversalisPas encore d'évaluation
- L'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)D'EverandL'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)Évaluation : 4 sur 5 étoiles4/5 (3032)
- Physique quantique pour les débutants: Découvrez les fondements de la mécanique quantique et la façon dont elle affecte le monde dans lequel nous vivons à travers ses théories les plus célèbresD'EverandPhysique quantique pour les débutants: Découvrez les fondements de la mécanique quantique et la façon dont elle affecte le monde dans lequel nous vivons à travers ses théories les plus célèbresÉvaluation : 5 sur 5 étoiles5/5 (2)
- L'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaD'EverandL'Art de La Magie au Bougie Wicca: Le Guide du Débutant à la Pratique de la Magie au Bougie de WiccaÉvaluation : 3 sur 5 étoiles3/5 (1)
- Les 10 Secrets pour une Vie Plus Heureuse avec la Maladie de ParkinsonD'EverandLes 10 Secrets pour une Vie Plus Heureuse avec la Maladie de ParkinsonPas encore d'évaluation
- Mes inventions (Traduit): Autobiographie de Nikola TeslaD'EverandMes inventions (Traduit): Autobiographie de Nikola TeslaÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)