Vous aimerez peut-être aussi
- Thermodynamique Fondements Et ApplicationsDocument573 pagesThermodynamique Fondements Et ApplicationsLamnister Zonon100% (3)
- DUNOD - Le Cours de Physique de Feynman - Mécanique 1 - R. FeynmanDocument362 pagesDUNOD - Le Cours de Physique de Feynman - Mécanique 1 - R. FeynmanJB_caesar100% (2)
- Cours de Chimie OrganiqueDocument114 pagesCours de Chimie Organiqueosefresistance100% (1)
- Photosynthèse: Les Grands Articles d'UniversalisD'EverandPhotosynthèse: Les Grands Articles d'UniversalisPas encore d'évaluation
- Présentataion+d avancement+de+ma+thèse+à+la+FS+30+01+2008Document23 pagesPrésentataion+d avancement+de+ma+thèse+à+la+FS+30+01+2008Farid Bou75% (4)
- Processus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisD'EverandProcessus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisPas encore d'évaluation
- CinetiqueDocument50 pagesCinetiquesidi mohamed el amine nekkalPas encore d'évaluation
- Cours Complet 1ere Chap13Document28 pagesCours Complet 1ere Chap13phytanjaPas encore d'évaluation
- Bioélectronique Concepts de Base (József ORSZÁGH)Document11 pagesBioélectronique Concepts de Base (József ORSZÁGH)GérardPas encore d'évaluation
- Réactivité Chimique: S3 Mip, FST - Fès Année Universitaire 2020/2021Document63 pagesRéactivité Chimique: S3 Mip, FST - Fès Année Universitaire 2020/2021Oumayma IkhlefPas encore d'évaluation
- Chimie GénéraleDocument175 pagesChimie GénéraleBopePas encore d'évaluation
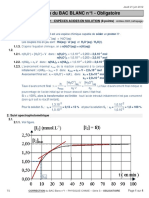
- TS - BAC Blanc N°1 Corrigé - ObligatoireDocument4 pagesTS - BAC Blanc N°1 Corrigé - ObligatoirephytanjaPas encore d'évaluation
- COURS CHIMIE GENERALE G1 POLYTECH 2015 - CopieDocument189 pagesCOURS CHIMIE GENERALE G1 POLYTECH 2015 - CopieAbel LetshuPas encore d'évaluation
- Orient DBDocument27 pagesOrient DBKlaus ERBAKPas encore d'évaluation
- Thermodynamique - WikipédiaDocument15 pagesThermodynamique - WikipédiaAdama NacanaboPas encore d'évaluation
- 357 Modélisation Des Bioréacteurs Morchain Première PartieDocument4 pages357 Modélisation Des Bioréacteurs Morchain Première Partieyoussef CHAFAIPas encore d'évaluation
- Cinétique ChimiqueDocument10 pagesCinétique Chimiquearfpower100% (2)
- Thermodynamique Et Thermique Chapitre IDocument9 pagesThermodynamique Et Thermique Chapitre ILove-God Ronzi100% (1)
- Cinétique ChimiqueDocument7 pagesCinétique ChimiquehajPas encore d'évaluation
- Ch9 Temps Evolution ChimiqueDocument4 pagesCh9 Temps Evolution Chimiqueالغزيزال الحسن EL GHZIZAL HassanePas encore d'évaluation
- Sommaire SilicateDocument184 pagesSommaire SilicateDjamila DadaPas encore d'évaluation
- Kermen - Isabelle ARDIST2005Document9 pagesKermen - Isabelle ARDIST2005Youssef AouadiPas encore d'évaluation
- CH1-historique Atome PDFDocument63 pagesCH1-historique Atome PDFRahme NormanPas encore d'évaluation
- Evolution SpontanéeDocument3 pagesEvolution SpontanéeNoury AbkPas encore d'évaluation
- DiffusionDocument10 pagesDiffusionrahil sabahPas encore d'évaluation
- Chap 2 Rappel de Cinétique Chimique Stœchiométrie Et Notion D'avancementDocument14 pagesChap 2 Rappel de Cinétique Chimique Stœchiométrie Et Notion D'avancementAPas encore d'évaluation
- L'hylémorphisme Dans Le Monde InorganiqueDocument24 pagesL'hylémorphisme Dans Le Monde InorganiqueAlexandre MazetPas encore d'évaluation
- Loi D'arrhenius Dans Le Vieillissement Des PolymèresDocument44 pagesLoi D'arrhenius Dans Le Vieillissement Des PolymèresAyoub DjouabiPas encore d'évaluation
- CatalyseDocument4 pagesCatalyseRoudaina BenzeguirPas encore d'évaluation
- Cours Cinéique UM6P 20 - 21Document57 pagesCours Cinéique UM6P 20 - 21Hifdi AyaPas encore d'évaluation
- Avogadro, Sa Constante Entre Mythe Et RéalitéDocument5 pagesAvogadro, Sa Constante Entre Mythe Et Réalitéالغزيزال الحسن EL GHZIZAL HassanePas encore d'évaluation
- Metathese AC 2004 273 Mars Astruc p1 3 PDFDocument9 pagesMetathese AC 2004 273 Mars Astruc p1 3 PDFJuan Jose SossaPas encore d'évaluation
- 2004 273 Mars Astruc p.3Document9 pages2004 273 Mars Astruc p.3Karfala KandePas encore d'évaluation
- Chap 01-Cinétique-Durée-RéactionDocument5 pagesChap 01-Cinétique-Durée-Réactionmube 75Pas encore d'évaluation
- Cours Thermodynamique P222 BCG S2Document109 pagesCours Thermodynamique P222 BCG S2ïLÿãsPas encore d'évaluation
- TSP2SP2Ch9T2 Resume CinetiqueDocument2 pagesTSP2SP2Ch9T2 Resume Cinetiqueallali hanaaPas encore d'évaluation
- Cours Chimie Ensa 2013 1Document55 pagesCours Chimie Ensa 2013 1Nawal ZakriPas encore d'évaluation
- Révisions Généralités Mécanique Fluides Méca Flu FicheDocument46 pagesRévisions Généralités Mécanique Fluides Méca Flu Fichebessama84Pas encore d'évaluation
- Chapitre 3 - CatalyseDocument14 pagesChapitre 3 - CatalyseKevin RavelondrambalaPas encore d'évaluation
- Ch3 Cinetique Et Catalyse ElDocument6 pagesCh3 Cinetique Et Catalyse ElNlem NdongoPas encore d'évaluation
- Correction To CinetiqueDocument5 pagesCorrection To CinetiquejaywalkPas encore d'évaluation
- Chapitre1 Biophysique PDFDocument9 pagesChapitre1 Biophysique PDFazouzrouaida49Pas encore d'évaluation
- ChC1 - Effets ThermiquesDocument16 pagesChC1 - Effets ThermiquesMamadou Alpha JallowPas encore d'évaluation
- Chimie Generale EUF1 - S1Document102 pagesChimie Generale EUF1 - S1Max GerbierPas encore d'évaluation
- Cours c9 - Tss 2019Document2 pagesCours c9 - Tss 2019mohamed laghribPas encore d'évaluation
- 6 Mecani 2015Document19 pages6 Mecani 2015Zizou LeePas encore d'évaluation
- Fom-Mr L2 PCDocument56 pagesFom-Mr L2 PCBaroka julien YANEPas encore d'évaluation
- Support de Cours CHM 122, S2, 2021-2022Document63 pagesSupport de Cours CHM 122, S2, 2021-2022Marie josé Edjongolo AkonoPas encore d'évaluation
- CCH6 CoursDocument2 pagesCCH6 CoursEL MEHDI EL HAMDOUCHIPas encore d'évaluation
- Cinétique ChimiqueDocument15 pagesCinétique ChimiqueYounes JaaidaniPas encore d'évaluation
- Chimie L1 MPCIDocument97 pagesChimie L1 MPCIAhmad CisséPas encore d'évaluation
- Artículo Modelado Transferencia de OxígenoDocument7 pagesArtículo Modelado Transferencia de OxígenoYoel Alfonso AcostaPas encore d'évaluation
- Suivi Temporel D'une Transformation ChimiqueDocument10 pagesSuivi Temporel D'une Transformation ChimiqueMhamed LaihemPas encore d'évaluation
- Cours ChimieDocument134 pagesCours ChimieMehdi Hajji100% (2)
- II 04 CinetiqueDocument3 pagesII 04 CinetiqueMohamed ElouakilPas encore d'évaluation
- Chapitre 1Document3 pagesChapitre 1idouiPas encore d'évaluation
- SPHM 1987 3 A1 0Document20 pagesSPHM 1987 3 A1 0Van FoliviPas encore d'évaluation
- Analyse 3Document19 pagesAnalyse 3ibrahimPas encore d'évaluation
- 1S.chim1.Cours - Quantite de Matiere Et Bilan de Matiere 06-07Document5 pages1S.chim1.Cours - Quantite de Matiere Et Bilan de Matiere 06-07Mohammed Ben AliPas encore d'évaluation
- Physique Statistique Hors D'équilibre - Processus Irréversibles Linéaires (PDFDrive)Document545 pagesPhysique Statistique Hors D'équilibre - Processus Irréversibles Linéaires (PDFDrive)Yannick DsprbsPas encore d'évaluation
- DynFluideM1 Cours PDFDocument204 pagesDynFluideM1 Cours PDFLahcen ElamraouiPas encore d'évaluation
- Thermodynamique-Chapitre 1-Description Macroscopique dÔÇÖun Syst+¿me Thermodynamique +á lÔÇÖ+®quilibreDocument22 pagesThermodynamique-Chapitre 1-Description Macroscopique dÔÇÖun Syst+¿me Thermodynamique +á lÔÇÖ+®quilibreKouame100% (1)
- La Cinétique ChimiqueDocument7 pagesLa Cinétique ChimiqueAS asPas encore d'évaluation
- Cours SM4001 PDFDocument105 pagesCours SM4001 PDFbentouamiPas encore d'évaluation
- Cinetique CDocument6 pagesCinetique CAyoub KhoyaPas encore d'évaluation
- DS1 Ondes Chap 1Document2 pagesDS1 Ondes Chap 1phytanja100% (1)
- Correc Devoir Ts 1Document4 pagesCorrec Devoir Ts 1phytanjaPas encore d'évaluation
- TS - DM1 - Phys 1-Chim 2Document2 pagesTS - DM1 - Phys 1-Chim 2phytanjaPas encore d'évaluation
- 2005 USA Correction Exo2 Ondes 5ptsDocument2 pages2005 USA Correction Exo2 Ondes 5ptsphytanjaPas encore d'évaluation
- 2-Activite - Theoreme EcDocument2 pages2-Activite - Theoreme EcphytanjaPas encore d'évaluation
- EX1C1 L'eau de Javel, Précaution D'emploi, StabilitéDocument1 pageEX1C1 L'eau de Javel, Précaution D'emploi, StabilitéphytanjaPas encore d'évaluation
- Cours VitesseDocument4 pagesCours VitessephytanjaPas encore d'évaluation
- devoirsurvTS n1Document5 pagesdevoirsurvTS n1phytanjaPas encore d'évaluation
- Sujetchimie Ensa 06Document6 pagesSujetchimie Ensa 06phytanjaPas encore d'évaluation
- Suivi TemporelDocument8 pagesSuivi TemporelphytanjaPas encore d'évaluation
- Exercice (Type Bac) Suivi Temporel D - Une Transformation ChimiqueDocument2 pagesExercice (Type Bac) Suivi Temporel D - Une Transformation ChimiquephytanjaPas encore d'évaluation
- Devoir Surveillé No 2Document2 pagesDevoir Surveillé No 2phytanjaPas encore d'évaluation
- Activités 3 Rotation Dun Solide Indéfor...Document5 pagesActivités 3 Rotation Dun Solide Indéfor...phytanjaPas encore d'évaluation
- Activités 3 Rotation Dun Solide Indéformable Autour D'un Axe FixeDocument5 pagesActivités 3 Rotation Dun Solide Indéformable Autour D'un Axe FixephytanjaPas encore d'évaluation
- Rotation D Un Solide Indeformable Autour D Un Axe Fixe CoursDocument10 pagesRotation D Un Solide Indeformable Autour D Un Axe Fixe Coursphytanja100% (1)
- Physique Chapitre2 Mouvement SolideDocument5 pagesPhysique Chapitre2 Mouvement SolidephytanjaPas encore d'évaluation
- Les Ondes Mécaniques ProgressivesDocument16 pagesLes Ondes Mécaniques ProgressivesphytanjaPas encore d'évaluation
- 7 Cours Du Moment de ForceDocument5 pages7 Cours Du Moment de Forcephytanja100% (1)
- TS - Chim 4 - TP Ch2Document2 pagesTS - Chim 4 - TP Ch2phytanjaPas encore d'évaluation
- TS - Phys 3 - TP Ph2 DiffractionDocument2 pagesTS - Phys 3 - TP Ph2 DiffractionphytanjaPas encore d'évaluation
- CoursExercices 01Document65 pagesCoursExercices 01phytanja100% (4)
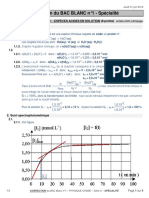
- TS - BAC Blanc N°1 Corrigé - Spécialité PDFDocument4 pagesTS - BAC Blanc N°1 Corrigé - Spécialité PDFphytanjaPas encore d'évaluation
- TS - BAC Blanc N°1 - ObligatoireDocument9 pagesTS - BAC Blanc N°1 - ObligatoirephytanjaPas encore d'évaluation
- TS - Phys 3 - Fiche Méthode - Figure de DiffractionDocument1 pageTS - Phys 3 - Fiche Méthode - Figure de DiffractionphytanjaPas encore d'évaluation
- TS - DM2 Corrigé - Phys 2-Chim 3Document3 pagesTS - DM2 Corrigé - Phys 2-Chim 3phytanjaPas encore d'évaluation
- TS - Chim 4 - ExosDocument2 pagesTS - Chim 4 - ExosphytanjaPas encore d'évaluation
- TS - DM 2 - Phys 2-Chim 3Document2 pagesTS - DM 2 - Phys 2-Chim 3phytanjaPas encore d'évaluation
- Cours3 IPM adressageIPDocument45 pagesCours3 IPM adressageIPAhmed MesaudaPas encore d'évaluation
- 'Etude Parasismique Selon L'eurocode 8Document10 pages'Etude Parasismique Selon L'eurocode 8Mohamed SEHLIPas encore d'évaluation
- DEVOIR STANDARDISE 4éme Mai 2022.Document2 pagesDEVOIR STANDARDISE 4éme Mai 2022.jean marie diedhiouPas encore d'évaluation
- Chapitre4 JqueryDocument14 pagesChapitre4 Jquerybendhifsaber22Pas encore d'évaluation
- Memoire FirewallDocument97 pagesMemoire FirewallLou LoulouPas encore d'évaluation
- Varaiteur 160 KW AltivarDocument8 pagesVaraiteur 160 KW AltivarsaidloubarPas encore d'évaluation
- Item 12 - ACE INDUS - Manuel Utilisateur Configuration SL7000Document13 pagesItem 12 - ACE INDUS - Manuel Utilisateur Configuration SL7000SALVADORPas encore d'évaluation
- Sa02 1112Document8 pagesSa02 1112Lawrence Mundene-timotheePas encore d'évaluation
- TS-chimie-C1-évolution Vers ÉquilibreDocument7 pagesTS-chimie-C1-évolution Vers Équilibrela physique selon le programme Français100% (2)
- Compléments AFCDocument12 pagesCompléments AFCMustapha ZianiPas encore d'évaluation
- Algorithmeknn 121213175830 Phpapp02Document14 pagesAlgorithmeknn 121213175830 Phpapp02SioudaPas encore d'évaluation
- BE Installation-et-deploiement-MyReport - BEDocument34 pagesBE Installation-et-deploiement-MyReport - BEDeyilon AubinPas encore d'évaluation
- Ana2 ch1Document9 pagesAna2 ch1ZaK SA-ïDPas encore d'évaluation
- 11 DenombrementDocument11 pages11 DenombrementMamane Issa TahirouPas encore d'évaluation
- Curriculum Vitae PDFDocument2 pagesCurriculum Vitae PDFromel tekamPas encore d'évaluation
- Base Et AcideDocument4 pagesBase Et AcideYves-donald MakoumbouPas encore d'évaluation
- AvantprojetDocument4 pagesAvantprojetwiam_alPas encore d'évaluation
- Cour Geotechnique Indice Des Vide Et AuteDocument5 pagesCour Geotechnique Indice Des Vide Et AuteCEDRIC SOBGUIPas encore d'évaluation
- SpeechDocument3 pagesSpeechsoufyane100% (1)
- Chapitre Ii Les Efforts InternesDocument27 pagesChapitre Ii Les Efforts InternesAYADI IMEDPas encore d'évaluation
- Cours BTS Puissance ElectromecaDocument11 pagesCours BTS Puissance ElectromecaAmadou SARRPas encore d'évaluation
- Atelier Asservissement CubotDocument25 pagesAtelier Asservissement CubotReda XiePas encore d'évaluation
- 543 Conception Et Schema Des Réseaux Fluidiques NDAME - ENGOLA - LEKANEDocument5 pages543 Conception Et Schema Des Réseaux Fluidiques NDAME - ENGOLA - LEKANEPaul Julbin NJOCKPas encore d'évaluation
- Cours Forces Pressantes Bep IndustrielDocument3 pagesCours Forces Pressantes Bep IndustrielFélix KouassiPas encore d'évaluation
- Modelisation de l'IPC Au SenegalDocument32 pagesModelisation de l'IPC Au SenegalASSELOKA AmadouPas encore d'évaluation
- Get StreamDocument11 pagesGet StreamStanescu MarianPas encore d'évaluation
- CoursinfoDocument52 pagesCoursinfontutaPas encore d'évaluation