Vous aimerez peut-être aussi
- Cours Chimie Ensa 2013 1Document55 pagesCours Chimie Ensa 2013 1Nawal ZakriPas encore d'évaluation
- Chimie GénéraleDocument175 pagesChimie GénéraleBopePas encore d'évaluation
- Kermen - Isabelle ARDIST2005Document9 pagesKermen - Isabelle ARDIST2005Youssef AouadiPas encore d'évaluation
- Chapitre 1 - I Generalite ThermodynamiqueDocument15 pagesChapitre 1 - I Generalite Thermodynamiqueayoub elhallaniPas encore d'évaluation
- Thermodynamique Et Thermique Chapitre IDocument9 pagesThermodynamique Et Thermique Chapitre ILove-God Ronzi100% (1)
- Thermodynamique II Chapitre 1Document72 pagesThermodynamique II Chapitre 1magiemots100% (1)
- COURS CHIMIE GENERALE G1 POLYTECH 2015 - CopieDocument189 pagesCOURS CHIMIE GENERALE G1 POLYTECH 2015 - CopieAbel LetshuPas encore d'évaluation
- BioénergétiqueDocument268 pagesBioénergétiqueAmi HBPas encore d'évaluation
- Cours Structure de La MatièreDocument82 pagesCours Structure de La MatièreCH BtissamPas encore d'évaluation
- Preprint Cinetique ACXIXmeDocument14 pagesPreprint Cinetique ACXIXmephytanjaPas encore d'évaluation
- Cours de Thermo - BIO141 - 24Document26 pagesCours de Thermo - BIO141 - 24kamalnjifenjou326Pas encore d'évaluation
- Cours Partie 1 Chimie en Solution AqDocument27 pagesCours Partie 1 Chimie en Solution AqchoroukbenhammouPas encore d'évaluation
- Introduction Sur La Thermodynamique Chimique Et Le Premier Principe. (Cours N°.1.)Document12 pagesIntroduction Sur La Thermodynamique Chimique Et Le Premier Principe. (Cours N°.1.)Rãfîk ZērkaňePas encore d'évaluation
- 1.biophysique1an-Solutions Applications Medicales2021-1Document39 pages1.biophysique1an-Solutions Applications Medicales2021-1Akram ChetouanePas encore d'évaluation
- chimie L1 MPCIDocument97 pageschimie L1 MPCIAhmad CisséPas encore d'évaluation
- Chapitre 1Document17 pagesChapitre 1yvesPas encore d'évaluation
- Thermo 1Document2 pagesThermo 1Raouan BijouPas encore d'évaluation
- Modélisation ThermodynamiqueDocument22 pagesModélisation ThermodynamiqueAmine Aitali100% (1)
- Prepa-Cours de Chimie-Usp-2023-2024-1Document46 pagesPrepa-Cours de Chimie-Usp-2023-2024-1namilohoahmedcoulibalyPas encore d'évaluation
- Cours ThermodyDocument54 pagesCours ThermodyHraiech IbtissemPas encore d'évaluation
- Expose Du Groupe KDocument8 pagesExpose Du Groupe KAbdoulaye SOUMAHPas encore d'évaluation
- Polycopié Thermodynamique IAP 2012 (Réparé)Document83 pagesPolycopié Thermodynamique IAP 2012 (Réparé)noured547244100% (1)
- 1 PrincipeDocument11 pages1 PrincipeM'barek BouzianiPas encore d'évaluation
- Chapitre I Généralités Sur Les Réactions en Solution Description D'un Système Et Évolution Vers Un État FinalDocument11 pagesChapitre I Généralités Sur Les Réactions en Solution Description D'un Système Et Évolution Vers Un État FinalMichel ZagoPas encore d'évaluation
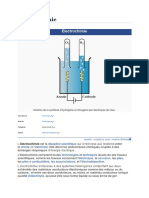
- ÉlectrochimieDocument6 pagesÉlectrochimieAmenPas encore d'évaluation
- Processus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisD'EverandProcessus irréversibles non linéaires en thermodynamique: Les Grands Articles d'UniversalisPas encore d'évaluation
- Bible 850 WordsDocument151 pagesBible 850 WordsAmam AmamPas encore d'évaluation
- Chapitre I Généralités Sur Les Réactions en Solution Description D'un Système Et Évolution Vers Un État FinalDocument11 pagesChapitre I Généralités Sur Les Réactions en Solution Description D'un Système Et Évolution Vers Un État FinalAlex N'zuePas encore d'évaluation
- Chapitre 1Document53 pagesChapitre 1Kawtar BahssainPas encore d'évaluation
- Thermodynamique - SMC4-part1 PDFDocument40 pagesThermodynamique - SMC4-part1 PDFAmine BriguichePas encore d'évaluation
- Cours ThermoDocument62 pagesCours ThermoJaphet DOSSOUPas encore d'évaluation
- Cours de Thermodynamique Chimique 2020 L2Document30 pagesCours de Thermodynamique Chimique 2020 L2Ismahane Ben100% (1)
- Cours de Thermodynamique Chimique 2020 L2Document46 pagesCours de Thermodynamique Chimique 2020 L2ayoub el houariPas encore d'évaluation
- Loi D'arrhenius Dans Le Vieillissement Des PolymèresDocument44 pagesLoi D'arrhenius Dans Le Vieillissement Des PolymèresAyoub DjouabiPas encore d'évaluation
- BioenergetiqueDocument50 pagesBioenergetiquemanelfor2004Pas encore d'évaluation
- L'hylémorphisme Dans Le Monde InorganiqueDocument24 pagesL'hylémorphisme Dans Le Monde InorganiqueAlexandre MazetPas encore d'évaluation
- Chapitre 1 - Les Systèmes ThermodynamiquesDocument13 pagesChapitre 1 - Les Systèmes ThermodynamiquesGhofrane benhamedPas encore d'évaluation
- Cours Thermodynamique ChimiqueDocument73 pagesCours Thermodynamique Chimiquerekik hibaPas encore d'évaluation
- La Matière Liquide Et Gazeuse Autour de Nous Contient Donc Une Immense Quantité DDocument2 pagesLa Matière Liquide Et Gazeuse Autour de Nous Contient Donc Une Immense Quantité Dfifisisi878Pas encore d'évaluation
- BIOENERGETIQUE CorrigéeDocument48 pagesBIOENERGETIQUE Corrigéesafemind100% (27)
- Le Concept de La Bioénergétique 2020 2021-ConvertiDocument10 pagesLe Concept de La Bioénergétique 2020 2021-ConvertiJacques Ahishakiye100% (1)
- Chapitre 2Document13 pagesChapitre 2Junias PakiPas encore d'évaluation
- Cours Thermochimie 2021Document109 pagesCours Thermochimie 2021Hiba AmalouPas encore d'évaluation
- American University of Kinshasa - ChigenDocument23 pagesAmerican University of Kinshasa - ChigengrandedamePas encore d'évaluation
- Exnicn R-Eueral D': RngenicurDocument39 pagesExnicn R-Eueral D': RngenicurAnis SELLAMPas encore d'évaluation
- Chapitre 1Document15 pagesChapitre 1ferdinandPas encore d'évaluation
- CHM108 Définitions Et Notions de BaseDocument5 pagesCHM108 Définitions Et Notions de BaseSam LessiPas encore d'évaluation
- 5393245Document62 pages5393245Soufiane OurrakPas encore d'évaluation
- Cours de ThermodynamiqueDocument101 pagesCours de ThermodynamiqueMama Samba SALLPas encore d'évaluation
- PolyphénolsDocument20 pagesPolyphénolsAmeni Ben nacerPas encore d'évaluation
- Introduction à la physique de la matièreD'EverandIntroduction à la physique de la matièreÉvaluation : 3 sur 5 étoiles3/5 (1)
- Ecoulements Mono Et Di-PhasiquesDocument162 pagesEcoulements Mono Et Di-PhasiquesAgent 47Pas encore d'évaluation
- Projet de Physique P6 STPI - P6 - 2013 28. Ailettes Thermiques. Etudiants - Enseignant-Responsable Du Projet - Diane DUVALDocument28 pagesProjet de Physique P6 STPI - P6 - 2013 28. Ailettes Thermiques. Etudiants - Enseignant-Responsable Du Projet - Diane DUVALjbouguechalPas encore d'évaluation
- DC ThermoDocument1 pageDC ThermoayadiPas encore d'évaluation
- Cours4 Les Reactions Chimie Organique Part1Document29 pagesCours4 Les Reactions Chimie Organique Part1Kalosoiretrotchgmail.com KalosoPas encore d'évaluation
- Cours 1 CHIMIE Nouveau ProgrammeDocument37 pagesCours 1 CHIMIE Nouveau Programmeabdelhadibens27Pas encore d'évaluation
- BiophysiqueDocument7 pagesBiophysiquealmnaouarPas encore d'évaluation
- Cours - BENCHIKH Abdelkader - Cours Et Exercices de ThermodynamiqueDocument108 pagesCours - BENCHIKH Abdelkader - Cours Et Exercices de Thermodynamiquekernoumehdi17Pas encore d'évaluation
- Cours - Thermodynamique 1Document70 pagesCours - Thermodynamique 1judeserruPas encore d'évaluation
- Faderco Spa Zi Les Eucalyptus ALGER, 99 16057 AlgeriaDocument13 pagesFaderco Spa Zi Les Eucalyptus ALGER, 99 16057 Algeriaأبو عبد البارئ الجزائريPas encore d'évaluation
- API20EDocument56 pagesAPI20EbenniaPas encore d'évaluation
- Ch4 Analyse SpectraleDocument9 pagesCh4 Analyse Spectraleالغزيزال الحسن EL GHZIZAL HassanePas encore d'évaluation
- Cours CNDDocument350 pagesCours CNDLéa BéquinPas encore d'évaluation
- Réacteurs Fluide Solide Catalytique (Partie D)Document6 pagesRéacteurs Fluide Solide Catalytique (Partie D)Siamo Quenn100% (2)
- AmidonDocument2 pagesAmidonlilian11100Pas encore d'évaluation
- Devoir Biochimie Med1Document2 pagesDevoir Biochimie Med1Aboubacar TraorePas encore d'évaluation
- M1577Document5 pagesM1577محمدلمينابراهيمالموريتانيPas encore d'évaluation
- Thèse Finale MMDocument161 pagesThèse Finale MMMOUAISSA Mohamed SalahPas encore d'évaluation
- Spécifications Des Produits PétroliersDocument5 pagesSpécifications Des Produits PétroliersFediMansouriPas encore d'évaluation
- Méthodes D'analyse ChromatographiqueDocument43 pagesMéthodes D'analyse ChromatographiquedesiréPas encore d'évaluation
- TP 4 LezoulDocument11 pagesTP 4 Lezoulcsdfs.chimiePas encore d'évaluation
- Catalogue GMT HALECO PDFDocument44 pagesCatalogue GMT HALECO PDFRABEBPas encore d'évaluation
- Stage Microalguesr PDFDocument32 pagesStage Microalguesr PDFcouvrefeuPas encore d'évaluation
- Pyrometallurgie Du Plomb - 220512 - 125033Document25 pagesPyrometallurgie Du Plomb - 220512 - 125033Viboys MupoyaPas encore d'évaluation
- TD de MicrobiologieDocument8 pagesTD de MicrobiologieAmel YahiaPas encore d'évaluation
- Brochure Mariseal FRDocument44 pagesBrochure Mariseal FRعتيقة يوسفيPas encore d'évaluation
- 2008 BTS Gemeau ET2 FR RéunionDocument14 pages2008 BTS Gemeau ET2 FR RéunionvodounnouPas encore d'évaluation
- Energie Nucleaire 2Document2 pagesEnergie Nucleaire 2KhairLatamnaPas encore d'évaluation
- Chapitre 3 Propriétés Des Verres 2022 PDFDocument18 pagesChapitre 3 Propriétés Des Verres 2022 PDFBtissam BelfassiPas encore d'évaluation
- m14 - Etude Du Tannage Vegetal Cuir-TanDocument41 pagesm14 - Etude Du Tannage Vegetal Cuir-TansamiPas encore d'évaluation
- Distillation1 PDFDocument52 pagesDistillation1 PDFfahim100% (1)
- Norme NF 206 CN P 19Document27 pagesNorme NF 206 CN P 19bou100% (1)
- TD ElectrochimieDocument2 pagesTD ElectrochimieYahia chenna100% (1)
- Méthodes de CalculDocument1 377 pagesMéthodes de CalculMEP EngineeringPas encore d'évaluation
- Polycope TP Enzymologie - 2021Document13 pagesPolycope TP Enzymologie - 2021a.elabbassiPas encore d'évaluation
- Transfert Technol Ferli Rosace165Document4 pagesTransfert Technol Ferli Rosace165chadlikamal1315Pas encore d'évaluation
- MSGM 3Document122 pagesMSGM 3maian saja100% (1)
- La Chimie Dans La CosmétiqueDocument46 pagesLa Chimie Dans La Cosmétiquejasserbarkouti00Pas encore d'évaluation
- TP 1Document6 pagesTP 1ISLAM DZPas encore d'évaluation