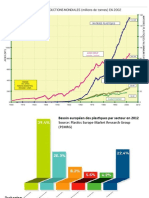
Vous aimerez peut-être aussi
- Polymères CoursDocument34 pagesPolymères CoursWallah Bakary100% (1)
- Chap 2 Macromolécules Partie 1Document5 pagesChap 2 Macromolécules Partie 1Hk EhPas encore d'évaluation
- Géometrie de MasseDocument10 pagesGéometrie de MasseKojak DoklPas encore d'évaluation
- Tableau Masses Molaires MoyennesDocument2 pagesTableau Masses Molaires MoyennesAbdou Djelama100% (1)
- Chapitre 1: Mécanique Rationnelle IIDocument88 pagesChapitre 1: Mécanique Rationnelle IIBodrick KahoziPas encore d'évaluation
- Cours 1 - TMDocument4 pagesCours 1 - TMLina alikhPas encore d'évaluation
- Chimie Des PolymèresDocument36 pagesChimie Des PolymèresRazvy100% (2)
- Cours Degradation Polymere N°3Document22 pagesCours Degradation Polymere N°3Ouahes HoudaifaPas encore d'évaluation
- Paramètre Structure PolymèreDocument11 pagesParamètre Structure PolymèreJeyani JEYARAJAHPas encore d'évaluation
- 2020 - Masse Molaire Moyenne PDFDocument1 page2020 - Masse Molaire Moyenne PDFAmélie Charleene ViennotPas encore d'évaluation
- Cours - Polymeres - CH4 - 2019-20120Document12 pagesCours - Polymeres - CH4 - 2019-20120KOUKI SOFIENPas encore d'évaluation
- Chapitre1 Transfert de MatiéreDocument14 pagesChapitre1 Transfert de MatiéreHa NahPas encore d'évaluation
- SRM Personnes 2019Document92 pagesSRM Personnes 2019LefevrePas encore d'évaluation
- Ch3.2.equilibre Liquide VapeurDocument5 pagesCh3.2.equilibre Liquide VapeurLounes RababPas encore d'évaluation
- Masses Molaires Moyennes Des PolymèresDocument19 pagesMasses Molaires Moyennes Des Polymèresachraf labiadPas encore d'évaluation
- Technique de transmission-chapII-Modulation D'amplitude AMDocument18 pagesTechnique de transmission-chapII-Modulation D'amplitude AMSouAi AzIzPas encore d'évaluation
- CV 1Document1 pageCV 1Imad Sisiniou RharboukiPas encore d'évaluation
- GueugneauL-2ème Leçon Do Et RéDocument2 pagesGueugneauL-2ème Leçon Do Et RéLuc GueugneauPas encore d'évaluation
- Devoir 1 Modele 8 Francais 1ac Semestre 2Document2 pagesDevoir 1 Modele 8 Francais 1ac Semestre 2الضحك حتى تصبح مضحك100% (1)
- PolymeresDocument8 pagesPolymeresCAPRIROCKPas encore d'évaluation
- Devoir 1college FR s2 Serie1Document2 pagesDevoir 1college FR s2 Serie1No ExpectationsPas encore d'évaluation
- 12 Intro Chimie Polymeres PDFDocument12 pages12 Intro Chimie Polymeres PDFfaslaPas encore d'évaluation
- Img 0003Document1 pageImg 0003Kouame Kouadjo SilverePas encore d'évaluation
- Article5 Courge-SistDocument19 pagesArticle5 Courge-SistZONGO100% (1)
- ThermoII ChapIIIDocument14 pagesThermoII ChapIII1442omisPas encore d'évaluation
- Chapitre Ii MefDocument8 pagesChapitre Ii MefJelly GiroudPas encore d'évaluation
- Cisse 3Document101 pagesCisse 3Samir Fassi FassiPas encore d'évaluation
- Polymères P1Document32 pagesPolymères P1Hadjer ZitounePas encore d'évaluation
- RRDFVDocument210 pagesRRDFVThelast SamauraiPas encore d'évaluation
- Le ParrainDocument1 pageLe ParrainRedouane Saioud100% (1)
- Correction TD 2Document4 pagesCorrection TD 2mahmoud bensassiPas encore d'évaluation
- L'apprenti - Pastouriau + Ah Les CrocodilesDocument1 pageL'apprenti - Pastouriau + Ah Les CrocodilesThibault DebehognePas encore d'évaluation
- Comptr Rendu Chimie MacroDocument4 pagesComptr Rendu Chimie MacroMehdi Jarrar100% (4)
- ENSP - Niveau2 - PHY228 - STATIQUE - Module 2 - LES TORSEURS UTILISES EN MECANIQUE - Cours - V22032020Document8 pagesENSP - Niveau2 - PHY228 - STATIQUE - Module 2 - LES TORSEURS UTILISES EN MECANIQUE - Cours - V22032020NANA EmmanuelPas encore d'évaluation
- PV L2 TEL 2017-2018-NormaleDocument4 pagesPV L2 TEL 2017-2018-NormaleYanis Diablo0% (1)
- Fransizca: Yabanci Dil BilgisiDocument28 pagesFransizca: Yabanci Dil Bilgisinsrlmd2Pas encore d'évaluation
- AAA123Document34 pagesAAA123Thelast SamauraiPas encore d'évaluation
- La Mole Unite de Quantite de Matiere Cours 1 2Document3 pagesLa Mole Unite de Quantite de Matiere Cours 1 2anissaPas encore d'évaluation
- CEDERTDocument132 pagesCEDERTThelast SamauraiPas encore d'évaluation
- Cours 1 - TM-1Document5 pagesCours 1 - TM-1Mål ÆkPas encore d'évaluation
- فرض الفصل الثاني - 2022-2021Document4 pagesفرض الفصل الثاني - 2022-2021lina AmokranePas encore d'évaluation
- Img 20210916 0002Document2 pagesImg 20210916 0002Kaled Lamouri100% (1)
- Best of Christophe Michalak (French Edition)Document104 pagesBest of Christophe Michalak (French Edition)Ryosuke Yagami100% (4)
- Plan Établi em - V2Document2 pagesPlan Établi em - V2etherwillPas encore d'évaluation
- CHAPIII MG MPI 20 21 Visio 06 01 2021Document14 pagesCHAPIII MG MPI 20 21 Visio 06 01 2021mahdi cherifPas encore d'évaluation
- RemerciementsDocument13 pagesRemerciementsThelast SamauraiPas encore d'évaluation
- Tégie Océan Bleu)Document1 pageTégie Océan Bleu)Thelast SamauraiPas encore d'évaluation
- Puissances Fiches Pedagogiques Maths 3AC PDF 2Document7 pagesPuissances Fiches Pedagogiques Maths 3AC PDF 2nassimlaacheloujPas encore d'évaluation
- Mathématiques PDFDocument304 pagesMathématiques PDFIsraelDiaz100% (1)
- AER2Document22 pagesAER2Thelast SamauraiPas encore d'évaluation
- TP Physique Compte Rendu Charge Et Decharge Dun CondensateurDocument12 pagesTP Physique Compte Rendu Charge Et Decharge Dun Condensateuranas habachiPas encore d'évaluation
- CH 3 Méthode de La Poussée Progressive (Pushover)Document9 pagesCH 3 Méthode de La Poussée Progressive (Pushover)Rayane Rana100% (1)
- PV-MI-Systèmes Des Télécommunications - 2017-2018-NormaleDocument2 pagesPV-MI-Systèmes Des Télécommunications - 2017-2018-NormaleYanis DiabloPas encore d'évaluation
- University of Cambridge International Examinations General Certificate of Education Ordinary LevelDocument12 pagesUniversity of Cambridge International Examinations General Certificate of Education Ordinary LevelVaishali CurumsingPas encore d'évaluation
- GraphiqueDocument3 pagesGraphiquekkbg05867Pas encore d'évaluation
- Series Numeriques ExercicesDocument2 pagesSeries Numeriques ExercicesomarPas encore d'évaluation
- AAA124ZDocument73 pagesAAA124ZThelast SamauraiPas encore d'évaluation
- Proposition D'investissement ABBEVILLE Bas Mesnil LMNP 3 LotsDocument21 pagesProposition D'investissement ABBEVILLE Bas Mesnil LMNP 3 LotskumacryptoinvestPas encore d'évaluation
- Boîte À Outils 03 Les VerresDocument16 pagesBoîte À Outils 03 Les VerresIkram GrPas encore d'évaluation
- IntroductionDocument1 pageIntroductionIkram GrPas encore d'évaluation
- Etude Par Résonnance Paramagnétique Électronique (RPE) de Polyméthacrylate de Méthyle (PMMA) Irradié Par Des Photons Gamma Issus de Cobalt 60Document75 pagesEtude Par Résonnance Paramagnétique Électronique (RPE) de Polyméthacrylate de Méthyle (PMMA) Irradié Par Des Photons Gamma Issus de Cobalt 60Ikram GrPas encore d'évaluation
- Finale Mémoire Et Bien OrganiserDocument84 pagesFinale Mémoire Et Bien OrganiserIkram GrPas encore d'évaluation
- Boîte À Outils 03 Les VerresDocument16 pagesBoîte À Outils 03 Les VerresIkram GrPas encore d'évaluation
- Azzi NadiaDocument66 pagesAzzi NadiaIkram GrPas encore d'évaluation
- Icm C#1Document31 pagesIcm C#1Ikram GrPas encore d'évaluation
- Caracterisation Des Masses MolairesDocument28 pagesCaracterisation Des Masses MolairesIkram GrPas encore d'évaluation
- Etude Thermique D Une Vitre en Film Composite de TypeDocument92 pagesEtude Thermique D Une Vitre en Film Composite de TypeArih FadiPas encore d'évaluation
- Caractérisation Par La Détermination Des Masses MoléculairesDocument23 pagesCaractérisation Par La Détermination Des Masses MoléculairesOualidPas encore d'évaluation
- Morphologie Et Propriétés Des PolymèresDocument10 pagesMorphologie Et Propriétés Des PolymèresIkram GrPas encore d'évaluation
- Math 3Document56 pagesMath 3Mounir DjlPas encore d'évaluation
- Cours 2Document14 pagesCours 2Ikram GrPas encore d'évaluation
- Cours 3Document22 pagesCours 3Ikram GrPas encore d'évaluation
- Cours #01 Méthodes D'analyse Des Matériaux 2022-2023Document13 pagesCours #01 Méthodes D'analyse Des Matériaux 2022-2023Ikram GrPas encore d'évaluation
- Cours #02 Méthodes D'analyse Des Matériaux Finalisé 2022-2023Document19 pagesCours #02 Méthodes D'analyse Des Matériaux Finalisé 2022-2023Ikram GrPas encore d'évaluation
- Cours 1Document13 pagesCours 1Ikram GrPas encore d'évaluation
- Etude de La Cinetique de La Reaction de Saponification de L'Acetate D'Ethyle Par Conductimetrie TP N°2Document2 pagesEtude de La Cinetique de La Reaction de Saponification de L'Acetate D'Ethyle Par Conductimetrie TP N°2Ikram GrPas encore d'évaluation
- Etude Cinetique D'Une Reaction Du 2 Ordre TP N°1Document2 pagesEtude Cinetique D'Une Reaction Du 2 Ordre TP N°1Ikram GrPas encore d'évaluation
- Rappel Cours: Spectrophotométrie Uv-VisibleDocument132 pagesRappel Cours: Spectrophotométrie Uv-VisibleIkram GrPas encore d'évaluation
- Math 3Document56 pagesMath 3Mounir DjlPas encore d'évaluation
- TD #9 Suite Corrigé 2018-2019Document4 pagesTD #9 Suite Corrigé 2018-2019Ikram GrPas encore d'évaluation
- Math 3Document56 pagesMath 3Mounir DjlPas encore d'évaluation
- Cours #02 Méthodes D'analyse Des Matériaux 2022-2023Document8 pagesCours #02 Méthodes D'analyse Des Matériaux 2022-2023Ikram GrPas encore d'évaluation
- Math 3Document56 pagesMath 3Mounir DjlPas encore d'évaluation
- Jadwal Genap 2223-2Document2 pagesJadwal Genap 2223-2nowo benyPas encore d'évaluation
- Chapitre 1 LES OUTILS MATHEMATIQUESDocument9 pagesChapitre 1 LES OUTILS MATHEMATIQUESa.ddPas encore d'évaluation
- Arval - Cofrastra 40Document16 pagesArval - Cofrastra 40helder.fradePas encore d'évaluation
- ExcisionDocument54 pagesExcisionAbdou Razak OuédraogoPas encore d'évaluation
- TP2Document4 pagesTP2Youssef Don RajawiPas encore d'évaluation
- Observons:: Nature Du Complément Circonstanciel de TempsDocument2 pagesObservons:: Nature Du Complément Circonstanciel de TempsMehdi YMPas encore d'évaluation
- 2nd - Exercices Corrigés - Variations D'une FonctDocument1 page2nd - Exercices Corrigés - Variations D'une Fonctalyahmed610Pas encore d'évaluation
- Memoire Inj Messaoud BENZOUAIDocument168 pagesMemoire Inj Messaoud BENZOUAIManong ShegueyPas encore d'évaluation
- Analyse D'une Situation de Communication en TaDocument2 pagesAnalyse D'une Situation de Communication en Taroger martin bassong batiigPas encore d'évaluation
- Tube VentouriDocument10 pagesTube VentouriMohammed BoulbairPas encore d'évaluation
- PV Liste-De-Prix Onduleurs HUAWEI 11-2023 FRDocument2 pagesPV Liste-De-Prix Onduleurs HUAWEI 11-2023 FRkoumbounisdimPas encore d'évaluation
- 3 Branches Triphasées en Injection Directe M215 Ou M250Document1 page3 Branches Triphasées en Injection Directe M215 Ou M250MbgardPas encore d'évaluation
- Controle Et Suivi Chantier RoutierhjhDocument14 pagesControle Et Suivi Chantier Routierhjhعثمان البريشيPas encore d'évaluation
- Prise en Main de Microsoft Office Excel 2016Document713 pagesPrise en Main de Microsoft Office Excel 2016max80% (5)
- Canalisations de Gaz NaturelDocument120 pagesCanalisations de Gaz NaturelJean-David DelordPas encore d'évaluation
- Introduction À La RobotiqueDocument19 pagesIntroduction À La RobotiqueRazzougui SarahPas encore d'évaluation
- Calendrier Des Examens Semestre Impair Janvier 2022 AlphaDocument28 pagesCalendrier Des Examens Semestre Impair Janvier 2022 AlphaMeg JustMegPas encore d'évaluation
- Caplp Externe Genie Electrique Electrotechnique Et Energie Epreuve 1 Doc RessourcesDocument28 pagesCaplp Externe Genie Electrique Electrotechnique Et Energie Epreuve 1 Doc RessourcesOus SàmàPas encore d'évaluation
- Depliant ELM MasterDocument3 pagesDepliant ELM MasterYazid AbouchihabeddinePas encore d'évaluation
- Format Eur FrancaiseDocument1 pageFormat Eur FrancaiseAdnan NandaPas encore d'évaluation
- Thèse Data IntegrityDocument83 pagesThèse Data IntegrityBasma YagoubiPas encore d'évaluation
- AnnexeDocument168 pagesAnnexeMoez AliPas encore d'évaluation
- TFE Gustave KISHATU MWAMBA Version Finale-1Document112 pagesTFE Gustave KISHATU MWAMBA Version Finale-1gustave kishatu100% (2)
- Les Étapes de Formation Des Roches SédimentaireDocument2 pagesLes Étapes de Formation Des Roches Sédimentairehamada2002Pas encore d'évaluation
- Marry Your Daughter Sheet - 1Document2 pagesMarry Your Daughter Sheet - 1Nurendung ZuliantoPas encore d'évaluation
- The Cuban Missile CrisisDocument8 pagesThe Cuban Missile Crisismilan.bodis523Pas encore d'évaluation
- 3 Partie Caractéristiques Des LubrifiantDocument32 pages3 Partie Caractéristiques Des Lubrifiantsamir belamriPas encore d'évaluation
- Racines Carrees BaseDocument8 pagesRacines Carrees Basejulien9562Pas encore d'évaluation
- Mesure de Compression Moteur Vers FinaleDocument46 pagesMesure de Compression Moteur Vers FinaleRaouf HarzallahPas encore d'évaluation
- Observatoire National de La Filiere Riz Du Burkina Faso (Onriz)Document6 pagesObservatoire National de La Filiere Riz Du Burkina Faso (Onriz)toni_yousf2418Pas encore d'évaluation
- Géobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainD'EverandGéobiologie de l'habitat et Géobiologie sacrée: Pour un lieu sainÉvaluation : 4.5 sur 5 étoiles4.5/5 (2)
- Harmonisation Energétique des Personnes: Manuel de Curothérapie 2020D'EverandHarmonisation Energétique des Personnes: Manuel de Curothérapie 2020Évaluation : 4 sur 5 étoiles4/5 (8)
- Technologie automobile: Les Grands Articles d'UniversalisD'EverandTechnologie automobile: Les Grands Articles d'UniversalisPas encore d'évaluation
- Manuel pour les débutants Fabriquez des savons naturelsD'EverandManuel pour les débutants Fabriquez des savons naturelsÉvaluation : 3 sur 5 étoiles3/5 (2)
- Améliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesD'EverandAméliorer votre mémoire: Un Guide pour l'augmentation de la puissance du cerveau, utilisant des techniques et méthodesÉvaluation : 5 sur 5 étoiles5/5 (2)
- Conception & Modélisation CAO: Le guide ultime du débutantD'EverandConception & Modélisation CAO: Le guide ultime du débutantPas encore d'évaluation
- Semer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumeD'EverandSemer avec succès pour rassembler avec abundance. Jardin organique et synergique: Calcul des meilleurs jours pour l'ensemencement de chaque légumePas encore d'évaluation
- Secrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieD'EverandSecrets ancestraux d'un maître guérisseur: Un sceptique occidental, un maître oriental et les plus grands secrets de la vieÉvaluation : 5 sur 5 étoiles5/5 (2)
- Le B.A.-Ba de la communication: Comment convaincre, informer, séduire ?D'EverandLe B.A.-Ba de la communication: Comment convaincre, informer, séduire ?Évaluation : 3 sur 5 étoiles3/5 (1)
- L'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)D'EverandL'Art de la guerre: Traité de stratégie en 13 chapitres (texte intégral)Évaluation : 4 sur 5 étoiles4/5 (3032)
- Transformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitD'EverandTransformez votre vie: Utilisez le pouvoir créateur qui est en vous pour construire votre vie à l'image de ce que vous voulez qu'elle soitÉvaluation : 4 sur 5 étoiles4/5 (14)
- Harmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020D'EverandHarmonisation Energétique des Lieux: Habitat et haut-lieux sacrés 2020Évaluation : 2.5 sur 5 étoiles2.5/5 (3)
- La vie des abeilles: Prix Nobel de littératureD'EverandLa vie des abeilles: Prix Nobel de littératureÉvaluation : 4 sur 5 étoiles4/5 (41)
- 20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsD'Everand20 Véritables remèdes de nos grands-mères pour maigrir vite et enfin perdre du poidsÉvaluation : 5 sur 5 étoiles5/5 (1)
- Histoire de la psychologie scientifique: De la naissance de la psychologie à la neuropsychologie et aux champs d'application les plus actuelsD'EverandHistoire de la psychologie scientifique: De la naissance de la psychologie à la neuropsychologie et aux champs d'application les plus actuelsPas encore d'évaluation
- Géologie de l'Amérique: Les Grands Articles d'UniversalisD'EverandGéologie de l'Amérique: Les Grands Articles d'UniversalisPas encore d'évaluation
- Jus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTD'EverandJus de Fruits et de Légumes Crus: 57 recettes faciles et un Guide Pratique Complet pour améliorer votre alimentation .: Santé, Vitalité et Minceur, avec ... ET DURABLEMENTPas encore d'évaluation
- 500 secrets pour avoir un potager merveilleuxD'Everand500 secrets pour avoir un potager merveilleuxÉvaluation : 2 sur 5 étoiles2/5 (1)
- Anatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursD'EverandAnatomie & 100 étirements essentiels: Techniques, Bénéfices attendus, Précautions à prendre, Conseils, Tableaux de séries, DouleursPas encore d'évaluation
- L'Ombre à l'Univers: La structure des particules élémentaires XIIfD'EverandL'Ombre à l'Univers: La structure des particules élémentaires XIIfPas encore d'évaluation
- Approvisionnement et traitement de l’eau: Les Grands Articles d'UniversalisD'EverandApprovisionnement et traitement de l’eau: Les Grands Articles d'UniversalisPas encore d'évaluation